FOOD INTAKE REGULATION AND OBESITY: HOMEOSTATIC AND HEDONIC CONTROL
Obesity is a multifactorial condition influenced by genetic, endocrine-metabolic, environmental and psychological factors. A delicate balance between three main biochemical and behavioral processes maintains body weight: food intake, energy expenditure control and energy storage control.1
Regulation of food intake by the Central Nervous System (CNS) depends on the interaction of a homeostatic component that aims the balance between energy and nutrients, and a hedonic component, which seeks food-associate pleasure (Figure 1).
Figure 1: Homeostatic and hedonic control of energy balance
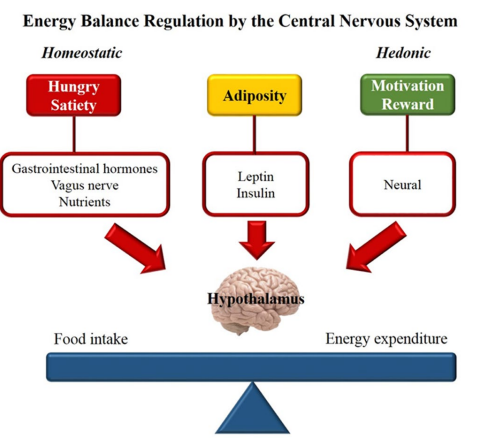
Homeostatic control of intake depends on the hormonal peripheral signaling produced in response to changes in nutrient concentrations. Leptin and insulin are the main hormonal adiposity signals, and by reaching the CNS trigger mechanisms that promote inhibition of food intake and increased energy expenditure.2 On the other hand, hunger and satiety sensations are communicated to the CNS by gastrointestinal hormones. During prolonged fasting, the stomach produces ghrelin that acts on the hypothalamus as an orexigenic signal. After food intake, ghrelin concentration falls, giving rise to the secretion of anorectic hormones, such as cholecystokinin (CCK), peptide YY (PYY) and glucagon-like peptide-1 (GLP1).3,4
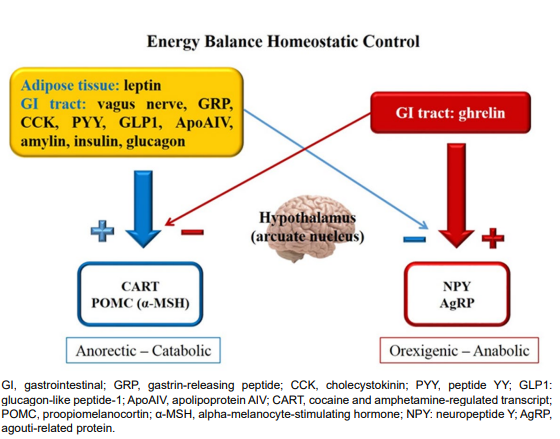
The main targets of peripheral adiposity, hunger and satiety signaling are neurons in the hypothalamus arcuate nucleus (ARC). In this nucleus there are orexigenic neurons that produce neuropeptide Y (NPY) and agouti-related protein (AgRP), in addition to anorectic neurons producing cocaine and amphetamine-regulated transcript (CART) and proopiomelanocortin (POMC), precursor of alpha-melanocyte-stimulating hormone (α-MSH) by the action of prohormone convertase 1 (PC1).1,5 The melanocortin 4 receptor (MC4R) plays an important role in the intricate hypothalamic appetite control. When leptin binds to its receptor on POMC neurons, α-MSH binds to MC4R, which produces a satiety signal. On the other hand, binding of AgRP to MC4R promotes increased intake.2,6 Leptin activates the POMC neurons and inhibits AgRP neurons.2,6,7 A scheme on the interaction of homeostatic intake control is depicted in Figure 2.
Figure 2: Homeostatic regulation of energy balance.
In humans, nutrition has not only physiological, but also social and behavioral roles. Food hedonic value is influenced by taste and previous experiences.4 Intake of highly palatable foods (high in sugar and fat) is able to “deregulate” appetite homeostatic control, perpetuating the stimulus to eat, which causes the intake to be primarily mediated by hedonic and not homeostatic needs.7 For decisions on food search, initiation and termination of meal to be properly taken, it is necessary the right integration between hypothalamic signals and cortical centers where substances such as opioids, endocannabinoids, gamma-aminobutyric acid (GABA), serotonin and dopamine (DA) act on the mechanisms of motivation and reward.2
Endogenous opioids as β-endorphin, enkephalin and dinorphin activate receptors in the accubens nucleus (NAc) disinhibiting orexigenic neurons in the Lateral Hypothalamic Area (LHA). Endocannabinoids impair leptin signaling, and interact with dopaminergic and opioid systems through the activation of CB1 receptors that inhibit melanocortin pathway.2,7
Only the role of eating facilitator through its action on NPY neurons and consequent blockage of POMC transmission was attributed to GABA.8 It is now known that GABA released by AgRP neurons is necessary to maintain a minimum level of appetite and the normal regulation of energy balance.9
Serotonin promotes satiety by acting directly in the ARC neurons, activating POMC and inhibiting AgRP neurons. It also inhibits orexins-producing neurons in LHA.2
DA is a catecholamine precursor of noradrenaline and adrenaline and is an endogenous neurotransmitter that modulates a number of physiological functions, including behavior, ion transport, vascular tone and blood pressure. Several experimental studies established DA as the main neurotransmitter of the reward system.10 It is currently considered the “pleasure molecule” or the “anti-stress molecule.” Previc11 stablished the concept of “dopaminergic society” and affirms that high DA concentrations was part of a general physiological adaptation to the increased meat consumption occurred two million years ago. The theory says that the “dopaminergic society” is characterized by high intelligence, personal destiny sense, religious/cosmic concerns and obsession with achieving goals.11
Regarding appetite, DA has varying effects depending on the brain area and the type of receptor stimulated. It has anorectic effect when it operates in the ARC, LHA and NAc, but acts as orexigenic in the ventromedial hypothalamic nucleus (VMH).2 Several studies have related the dopaminergic brain circuits in eating behavior.7 Animal studies reveal that the consumption of high-sugar or high-fat meals promotes the DA release in NAc.7,12 The intake of a tasty meal for humans induces the DA release in a magnitude proportional to the meal degree of pleasure.13
OBESITY AS PART OF THE “REWARD DEFICIENCY SYNDROME – RDS”
Structures and cortico-limbic-striatal circuits form the brain reward system. Pleasurable stimuli activate this system and lead the individual to seek positive reinforcement of every type, not only food.14 The brain reward cascade starts in the hypothalamus, where serotonin acts as a neurotransmitter stimulating the enkephalin release, which in turn inhibits GABAergic neurons in the substantia nigra. These GABAergic neurons act in the fine adjustment of DA amount that will be released in the NAc, the brain reward site (Figure 3).10
Figure 3: Interaction of various neurotransmitters that forms the “Brain Reward Cascade”. Adapted from Blum et al10
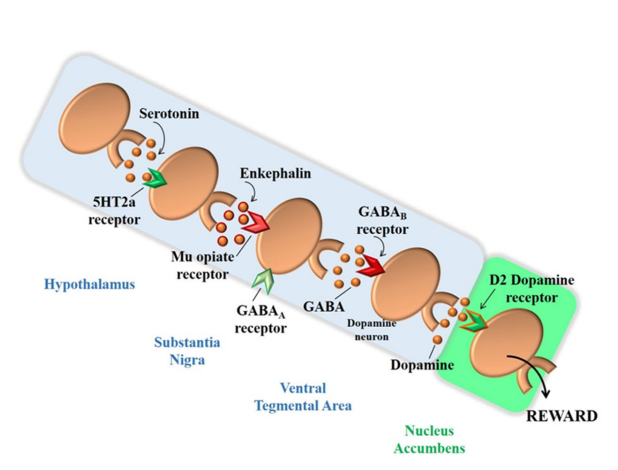
Studies show that low brain DA concentrations relate to greater vulnerability to substance abuse and abnormal behavior. It is known that all addictive drugs, as well as gambling, sex, food and even music promote DA release in the brain reward site.10 In 1996, the term “Reward Deficiency Syndrome – RDS” was stablished, in order to define hypodopaminergic states-associated behaviors, which predispose to obsessive-compulsive behaviors and to impulsiveness.15 The following changes are included in the RDS: a) Addictive behaviors: alcoholism, multiple substances abuse, obesity, smoking; b) Impulsive behaviors: attention deficit hyperactivity disorder (ADHD), Tourette’s syndrome, autism; c) Compulsive behaviors: abnormal sexual behavior, addiction to gambling and betting; d) Personality disorders: conduct disorder, antisocial personality, aggressive behavior and generalized anxiety.15
GENETIC FACTORS RELATED TO OBESITY
Common obesity, also named exogenous obesity, is a complex disease with multifactorial etiology. Pleiotropic genetic syndromes and monogenic diseases account for only 1% of obesity cases.16,17 The most common forms of monogenic obesity occur due to mutations in genes related to hypothalamic control system of energy balance, as leptin-melanocortin system, which result in changes in the concentration and/or activity of hormones, receptors and enzymes, including leptin and its receptor, POMC, MCR4 and PC1.18 In addition, there may be mutations in genes affecting the hypothalamus development and therefore, promoting obesity.6 It is also important to note that obesity may be a central component of several pleiotropic syndromes, such as Alstrom, Albright, Pader-Willi, Bardet-Biedel, Fragile X, among other syndromes.17
In complex diseases such as common obesity, it is necessary that genetic factors are associated with a favorable environment for the phenotype emergence. The “thrifty genotype” hypothesis, described by Neel19 proposes that genetic variations that result in higher capacity to store energy as fat were positively selected in food deprivation times. It is believed that over thousands of years this “thrifty genotype” has perpetuated and was essential in mankind evolution. This theory suggests that genes included in the “thrifty genotype” are responsible for the great ability to accumulate energy as fat, the ability to save energy at critical periods, the capacity to “turn off” non-essential metabolic pathways and to facilitate the intake of large amounts of food whenever they are available.20 Currently, this same “thrifty genotype” has become disadvantageous, due to the easy access to energy-dense foods and to the low energy expenditure, which could explain the current obesity epidemic.
In 2007 Speakman published the “predation release” hypothesis, as an alternative to the “thrifty genotype” theory.21 Based on anthropological and epidemiological evidence, genetic screening and experimental research, the theory suggests that the greater skill of lean individuals selected those best adapted to the search for food and to escape from predators; until fire was discovered in the Paleolithic period, and there was a significant increase in body weight over time. The theory attributes this increase in weight not only to the cooking capacity and better palatability of foods, but mainly to the fact that the fire was able to keep out the main predators, reducing energy expenditure. It also suggests that the initial genetic network responsible for low weight and high body performance has been suppressed and lost over the millennia.20,21
Common forms of obesity result from the interaction between variations in different genes and a favorable environment. Generally, many studies suggest a strong genetic component in human obesity.22,23,24,25,26,27 Studies report that in response to low calorie diets, some individuals lose weight more easily than others, and those carrying the same genotype respond in a similar manner when exposed to the same diet. Researches with monozygotic twins show that heredity accounts for 40 to 70% of inter-individual variation in cases of common obesity.28 Differences between individuals and their predisposition to weight gain indicate that common variations in genomic DNA sequence, represented mainly by the Single Nucleotide Polymorphisms (SNP), may be responsible for the weight gain.27,29 However, despite its great importance, the search for genes that raise the risk for obesity has not been easy.28,30 It is still a challenge for the scientific community to separate the genetic component from the environmental one in the etiology of this disease. Individuals who are more susceptible to accumulating fat can carry risk variants in genes that influence appetite control (NPY, POMC, MC4R, etc.), cellular machinery regulation (FTO, DRD2, etc.), lipid metabolism and adipogenesis (PPAR, APOE, APOA1, PLIN, etc.), energy expenditure (UCP1, UCP2), insulin signaling (INSR, etc.) and inflammation (ADIPOQ, IL6, RETN, etc).18,27
The polygenic nature of common obesity makes the discovery of risk genes and their variants a challenging task. Different approaches have been developed to elucidate the genetic component of obesity, such as GWLS (Genome-Wide Linkage Studies), that include co-segregation studies of certain chromosomal regions with a trait or disease30; analysis of candidate genes involved in plausible physiological pathways; and GWAS (Genome-Wide Association Studies), that track markers throughout the genome to identify associated polymorphisms.28 Through a meta-analysis of 37 GWLS, Saunders and co-workers concluded that this is not an effective approach to identify genetic variants for common obesity, as they did not locate any locus with conclusive evidence.31
Studies on candidate genes intended to identify the relation between one or more polymorphisms and a phenotype. In obesity, genes involved in the regulation of food intake, energy expenditure, lipid and glucose metabolism and adipose tissue development have been studied. In addition, genes described in monogenic forms of obesity have been investigated for a possible role in the common obesity genesis.6 However, replication of most results has been somehow inconsistent, and so the findings of candidate gene studies remain obscure.32
In GWAS, unlike in the candidate gene approach, no assumption of the investigated gene function is made. These studies are based on the association of several markers, usually SNP and the approach is particularly useful in common complex diseases, such as obesity and diabetes.6
The latest update of “the human obesity gene map”, performed by Rankinen and co-workers associated obesity with 253 loci after analysis of 61 GWAS.33 The number of associations between SNPs and obesity has 127 candidate genes described, and of these, 22 genes are supported by more than five studies. The map shows loci in all chromosomes, except for the Y.33
A more recent meta-analysis from GIANT (The Genetic Investigation of ANthropometric Traits) Consortium conducted in adults34 established 32 loci of susceptibility to Body Mass Index (BMI), several of which were confirmed in French and German children with severe obesity.35 In 2015, 97 BMI-associated loci were identified in a GWAS with 339,224 subjects (~95.0% of European and ~5.0% of non-European descent) from 125 studies (82 with GWAS and 43 with Metabochip results). From those 97 loci, 56 were novel.36
Specifically for the childhood early-onset obesity, studies show that heredity is an important factor.37,38 A large study of the childhood obesity genetics evaluated 5,530 cases and 8,318 controls through the analysis of 14 scientific papers, and the strong genetic influence in the childhood obesity development was definitely verified.39 Three novel loci were identified in a meta-analysis with 47,541 children from 33 studies (discovery and replication phases).40
GENETIC VARIATIONS IN THE DOPAMINERGIC REWARD SYSTEM: DOPAMINE RECEPTOR D2 GENE (DRD2)
Taking into account that DA plays a crucial role in the brain reward circuit and is involved in food behavior, the study of genetic polymorphisms that affect the availability and secretion of DA has been standing out.
The DRD2 gene is located on chromosomal region 11q22-q23, with about 66,097 base pairs. This gene encodes the D2 subtype receptor, a transmembrane protein that couples to G proteins and inhibits the activity of adenylate cyclase. In an alternative splicing mechanism, the DRD2 encodes two molecularly distinct protein isoforms – D2S and D2L – which are co-expressed, although the D2L production is favored. These two isoforms differ by the presence of 29 additional amino acids at D2L.41 Both forms of D2 receptor have distinct physiological functions. The D2L acts mainly in postsynaptic regions while the D2S has a presynaptic self-receiving function.42 This gene was included in “the human obesity gene map” supported by five candidate genes studies performed only with adults.33
DRD2 TAQIA POLYMORPHISM
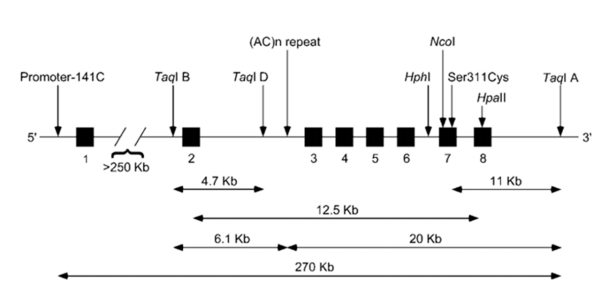
The DRD2 is highly polymorphic and there are already many SNPs cataloged and described (Figure 4). However, increasing attention has been given to C32806T SNP (rs1800497), characterized by the exchange of a cytosine for a thymine in a repetitive region in ANKK1 (gene codifying ankyrin), located downstream of the DRD2.43 This SNP is also known as TaqIA and appears to affect the D2 receptor availability. Variant allele A1 (T) is associated to a reduced metabolic rate of glucose in dopaminergic areas of the human brain, which indicates a low activity of dopaminergic neurons.44
Figure 4: DRD2 human gene with location of the most studied polymorphisms. Boxes represent exons and lines represent introns. Adapted from Noble, 2003.44
Variations in the expression and activity of dopaminergic receptors and in DA release are related to overeating and obesity. Mice with reduced density of DRD2 receptors in the striatum dorsal side, when fed with fat-rich diet gained more weight than those with normal density DRD2. The increase in DRD2 mRNA expression in the nucleus accumbens and in the putamen ventral part of obese mice, when compared with obesity-resistant mice, seems to be a compensatory response to the DRD2 pathway lower activation induced by overeating.45,46
Studies have suggested that obese individuals may have decreased DA availability through a mechanism of dopaminergic D2 receptors downregulation in the dorsal and lateral striatum.47,48,49 Drugs blocking D2 receptors increase the appetite, and those that raise central AD concentration have anorectic effects.48
In addition, researches with adult humans suggest that increases in body mass are associated with the DRD2 A1 allele.15,50,51 In studies with positron emission tomography, the A1 allele was associated with lower density of the DRD252 and with reduction of glucose metabolism in human brain dopaminergic regions.53 All RDS components, including obesity, were related to a low dopaminergic function due to the association with the DRD2 A1 allele.10,15,54
The SNP C32806T in DRD2 is also associated with a reduced dopaminergic brain activity55 and the A1 allele was initially associated with BMI increase.15,56 However, there are few studies28 verifying the association of this polymorphism in children and adolescents.
It has been observed large variation in the allelic frequencies regarding the DRD2 TaqIA SNP, even in populations of the same country. For example, in two studies with Turkish obese children the variant allele frequency was 51.0% in one57 and only 20.0% in the other research.58 In the Netherlands, the A1 allele frequency was 18.3% in obese children59 and in North-American studies with obese children, it ranged from 17.0%60 to 38.5%.61 In a Brazilian study with obese and normal weight (controls) children, the A1 allele frequencies were 34.5% and 23.0%, respectively, and a statistically significant association between the A1 allele and obesity was verified.62
FINAL REMARKS
Food intake is controlled by an interrelation between homeostatic and hedonic factors. The search for food-associated pleasure involves the same neuronal pathway that is stimulated by addictive/compulsive behaviors (alcohol, gambling, sex and drugs), the so called “hypothalamic reward system”, that ends in DA. In this context, the minor A1 allele of the DRD2 TaqIA polymorphisms has been associated with dopaminergic activity and increased obesity risk. Recognition of individuals predisposed to developing obesity through the determination of risk polymorphisms can guide prevention and treatment actions.
ACKNOWLEDGMENTS
We would like to thank Mr. Andre Hedlund for the English review.
AUTHOR’S CONTRIBUTIONS
RMP, CC and ADC wrote and approved the final manuscript.
CONFLICTS OF INTEREST (COI) STATEMENT
The authors declare no conflicts of interest.


