INTRODUCTION
Obesity is a serious and growing public health problem. It is a significant risk factor for the leading causes of mortality, including type 2 diabetes, cardiovascular diseases, and certain types of cancer. The number of individuals with weight excess (obesity+overweight) increased from 857 million in 1980 to 2.1 billion in 2013, and it is projected to reach levels of 89% and 85% of men and women, respectively, in 2030.1,2 Until recently, obesity was considered only as a consequence of an imbalance between intake and energy expenditure; obesity is now seen as a neurobehavioral disease, in which there are alterations in the hypothalamic control of hunger and satiety and energy expenditure.3,4
METHODS
We carried out a PubMed search, along with excerpta medica database (EMBASE)/Cochrane library, Web Sciences for the Medical Subject Headings (MeSH) Terms “obesity’’AND genetics” for the past 5-years.
RESULTS
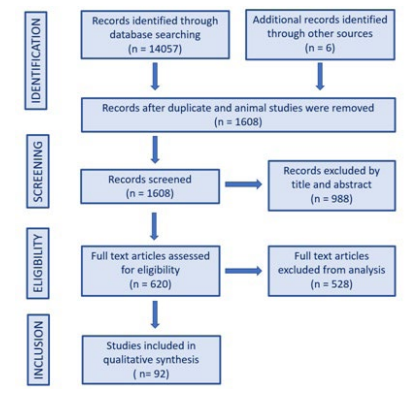
We found a total of 14057 articles pertaining to obesity and genetics together, after exclusions and getting articles searching through cross references, 92 articles were selected for qualitative synthesis, and 40 are the basis for this review (Figure 1).
Figure 1. Article Selection Flowchart
Obesity Epidemic
The obesity epidemic has been largely attributed to changes in lifestyle habits established over the past three decades. These changes are mainly attributed to excessive nutrition and decline in physical activity,5,6 as well as additional factors such as reduced intestinal microbiota diversity, sleep duration, endocrine disruptors, and reduced variability of the ambient temperature.5,7 However, the obesogenic environment is not sufficient to determine the presence of obesity, and it is necessary that the lifestyle becomes associated with a personal predisposition for the phenotype to emerge.8 Two main evolutionary hypotheses try to explain the current obesity epidemic. They are the “thrifty genotype” and “predator release” hypothesis.
Thrifty Genotype Hypothesis
The “thrifty genotype” hypothesis, described by Neel in 1962,9 proposes that genetic variations that result in a higher capacity to store energy as fat were positively selected in times of food deprivation. It is believed that over thousands of years this “thrifty genotype” has been perpetuated and was essential in the evolution of humanity. It is postulated that the genes that compose the “ thrifty genotype” are responsible for: higher capacity to accumulate energy in the form of fat, ability to save energy in critical periods, ability to “turn off” non-essential metabolic pathways and ability to ingest large amounts of food whenever these are available.10,11 The same “thrifty genotype” is currently disadvantageous because of easy access to high densely energetic foods and low caloric expenditure.9
Predator Release Hypothesis
In 2007, Speakman12 published the “predator release theory”, complementary to the “thrifty genotype” hypothesis.12 Based on anthropological and epidemiological evidence, genetic tracing and experimental research, the theory postulates that the higher agility characteristic of lean individuals has selected them who are better adapted for food search and escape from predators. That was true until the discovery of fire in the paleolithic period when it is observed a significant increase in body weight over time. The theory attributes this increase in weight not only to the cooking capacity (that leads to better palatability of food and greater absorption of nutrients), but fundamentally to the fact that fire keeps away the main predators (that leads to a reduction on energy expenditure on the run to escape from natural predators). The theory suggests that the initial genetic network responsible for low weight and high body performance characteristics has been suppressed and lost over the millennia.11
Genetic Predisposition to Obesity
Several pieces of research show the importance of genetics in the susceptibility to obesity. Studies with twins and adopted children show that 55 to 80% of the variation of body mass index (BMI) is attributed to genetic factors.13,14,15 The concordance rate for obesity is higher among monozygotic than dizygotic twins; the weight of adoptive children is closer to that of their biological parents than to their adoptive parents.13,14,15
According to the genetic criteria, obesity is classified as16:
A) Monogenic – when a mutated gene is responsible for the phenotype;
B) Syndromic – when a set of specific symptoms are present and a small group of genes is involved; usually the term is used to describe obese patients with cognitive delay, dysmorphic features, organ-specific abnormalities, hyperphagia, and/or other signs of hypothalamicdys function.
C) Polygenic – also called “common” obesity, present in up to 95% of cases. Many genes add up to provide a further risk to the individual, and if associated with some habits culminate in obesity.
Monogenic Obesity
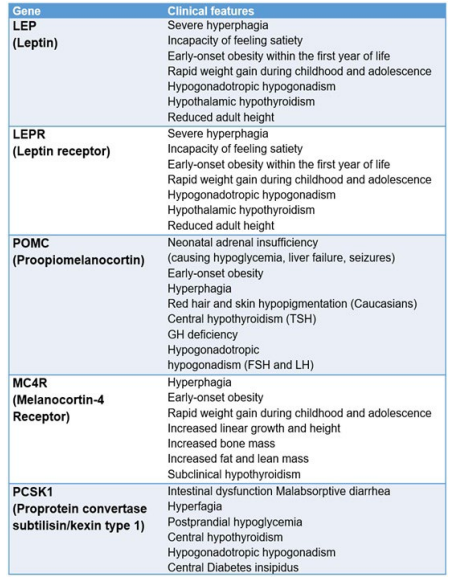
Monogenic obesity disorders are a heterogeneous group of rare conditions that increase food intake and reduce energy expenditure.16 Many occur from mutations in genes related to the hypothalamic system of energy balance control,8,11,16,17 as the leptin-melanocortin system. These mutations result in changes in the concentrations and/or activity of hormones, receptors and enzymes, leading to the phenotype of intense hyperphagia with early-onset obesity, sometimes associated with endocrine abnormalities.8,16,17,18 The main genes involved in monogenic obesity are summarized in Chart 1.
Chart 1. Monogenic forms of Obesity16
Syndromic Obesity
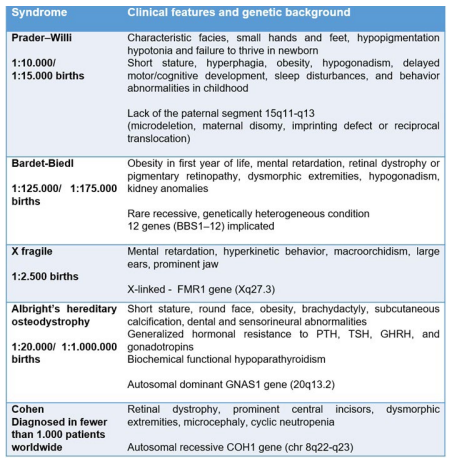
The term “syndromic obesity” refers to patients with early-onset obesity associated with an intellectual deficit, dysmorphic features, organ-specific abnormalities, extreme hyperphagia, and/or other signs of hypothalamic alteration.16,19,20 More than 100 syndromes associated with obesity were described; the most common are summarized in chart 2.17,21
Chart 2. Syndromic Obesity17,21
Polygenic Obesity
Polygenic obesity, also called common obesity, is the most prevalent type of obesity. It is multifactorial and depends not only on genetic factors but also on the existence of a favorable environment, established by an “obesogenic” lifestyle, with overfeeding, sedentary life, stress, among others. The genetic susceptibility comes from the cumulative effect of the contribution of several genes, with each presenting a slight effect on BMI, in a polygenic pattern.17,22
Differences between individuals and their predispositions to weight gain indicate that common variations of the genomic DNA sequence may be responsible for weight gain.19,23 However, in spite of its great relevance, the search for the genes that raise the risk for obesity has not been easy.24,25 It is still a challenge for the scientific community to separate the genetic element from the environmental component in the etiology of this disease. Individuals more susceptible to excessive adiposity may carry risk variants in the genes that influence appetite control (NPY, POMC, MC4R, etc), the regulation of cellular machinery (FTO, DRD2, etc), lipid metabolism and adipogenesis (PPAR, APOE, PLIN, etc.), energy expenditure (UCP), insulin signaling (IRS, etc) and inflammation (ADIPOQ, IL6, RETN, etc).18,23,26
Different approaches have been developed to elucidate the genetic component of polygenic obesity: Candidate gene, genome-wide linkage study (GWLS) and genome-wide association study (GWAS).
Candidate Gene
Association studies of candidate genes aim to identify the relationship between one or more polymorphisms and a phenotype. During the mid-1990s, studies began to identify common genetic variants (also called SNP – single nucleotide polymorphisms) that contribute to obesity susceptibility.27
The genes considered candidates were analyzed because of previous biochemical, physiological and/or clinical research, or even because of their location (in a region of linkage/association) or pharmacological findings, indicating a correlation with BMI variation; being performed in cases of extreme and early-onset obesity or in transgenic animal models.27,28 The search was then concentrated in those genes that play fundamental roles in the central or peripheral pathways of control of energy consumption and expenditure, influencing the regulation of food intake, energy expenditure, lipid and glucose metabolism, and adipose tissue development.28 Hundreds of genes have already been investigated as candidates to provide susceptibility to obesity, yet only a few have demonstrated a convincing association,27 and the replication of the results of most of the work has been inconsistent, so the conclusions of the candidate gene studies remain obscure.29
Genome Wide Linkage Study (GWLS)
In the late 1990s, the GWLS emerged. These studies have a generating hypotheses approach of certain chromosomal regions co-segregating with a trace or disease. They screen the entire genome of related individuals, with about 400-600 polymorphic markers, to identify chromosomal regions that segregate with obesity-related traits.27
Saunders et al,30 after conducting a meta-analysis of 37 GWLS studies, concluded that this is not an effective strategy for detecting genetic variants for common obesity since they did not locate any locus with convincing evidence.
Genome Wide Association Study (GWAS)
The GWAS, unlike previous approaches, proved to be quite efficient.28 In these studies, there is no assumption of the function of the gene being investigated. They are based on the association of several markers, special needs plans (SNP) usually, identifying genomic regions rather than specific genes, and are particularly useful in complex common diseases such as obesity and diabetes.5,28
The high success of this type of study stems from three factors: 1) the human genome is screened at a very high resolution with high-density scans, since more than two million genetic variants are tested for association with the characteristic of interest; 2) the sample size is much larger than in the linkage studies because participants do not need to be related, 3) the rigorous level of significance, established by the study design format, in two steps. The first stage identifies the loci for which the associations reach high-levels of significance in the genome scan, and then the second stage tests the loci for association in an independent series of samples. A locus is considered established when the association reaches a significance of <5×10-8 in the subsequent meta-analysis of the results of the first and second stages. Thus, the GWAS studies provide highly credible and robust association results.22,27,31
GWAS studies were important to understand the genetics involving obesity. Most studies were performed in white Europeans and addressed various loci in relation to BMI, body fat, waist and hip waist ratio, extreme and early-onset obesity.5,27 These studies evolved in 4 waves with a progressive increase of the sample.27
In the first wave, genetic variations were identified in the fat mass and obesity-associated protein (FTO) intron. The FTO is related, in addition to BMI, to the risk of obesity, abdominal circumference, body fat percentage and with childhood obesity. The second wave corroborated the association of obesity with FTO variations and identified a locus related to the MC4R. Mutations in MC4R are one of the causes of extreme obesity in childhood. The third and fourth waves identified new loci for BMI and, by the end of four waves; GWAS had identified 32 loci unequivocally associated with BMI.27
In 2006, Rankinen et al after evaluating 61 GWAS studies, updated the “genetic map of human obesity”, which at that time had 253 loci on all chromosomes except Y.32
In 2010, the giant-cell tumor medical definition (GIANT) consortium conducted in adults only33 established32 susceptibility loci for BMI, several of which were confirmed in French and German children with severe obesity.34 In 2012, the largest genome-wide association studies (GWAS) meta-analysis study was carried out on children: 5530 cases and 8318 controls were evaluated and the strong genetic influence on the development of childhood obesity was verified.35
In 2015, Locke et al36 published a GWAS study with approximately 340.000 individuals, identifying 97 loci with 2.1 million genetic variations, accounting for 2.7% of the BMI variation.36 Most of the seloci are expressed in the Central Nervous System (CNS) and carry genes involved in pathways that affect the neuro-circuits of appetite regulation and satiety (BDNF, MC4R and NEGR), as well as insulin secretion and action pathways (TCF7L2, IRS1), adipogenesis and energy and lipid metabolism (FTO, RPTOR, MAP2K5). Some genes also involved in monogenic non-syndromic obesity are associated with polygenic obesity and present common polymorphisms in PCSK1, MC4R, and POMC.8,36
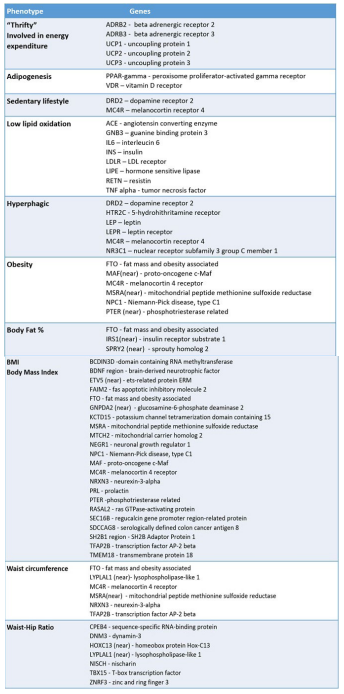
In 2019, Khera et al31 performed a metanalyses of GWAS studies and developed the first conclusive genetic risk score for obesity. After assessing 2.1 million SNPs in more than 300.000 individuals, the authors developed the genomic polygenic score (GPS) that allows the identification of people with high susceptibility to obesity. Each variant is individually associated with minimal differences in birth weight but predicts clear weight differences during early childhood and profound differences in weight trajectory and risk of developing severe obesity in subsequent years. Therefore, like GPS, other risk scores created from computational algorithms and large data sets are expected to identify a subgroup of the population that is at substantial risk for severe obesity in some cases equivalent to rare monogenic mutations and others that enjoy considerable protection.22,31 Chart 3 summarizes the phenotypes related to genes involved in the genesis of polygenic obesity.
Chart 3. Phenotypes and Genes Involved in Polygenic Obesity27,32
Epigenetics and Obesity
Epigenetics refers to changes that occur in deoxyribonucleic acid (DNA) that do not alter the sequence of nitrogenated bases but can control chromatin compaction and interfere with gene expression through the mechanisms of DNA methylation, histone modification, and gene silencing throw non-coding micro ribonucleic acid (mRNA) (small RNA molecules that bind to messenger RNA and there by block protein translation).37
The intrauterine environment and nutrient supply during the 1000 days (from conception to the second year of life) modulate the expression of genes involved in appetite regulation, insulin sensitivity, among others and can modify the risk of developing obesity and metabolic diseases.8,37,38,39 Exposure to maternal hyperglycemia in utero alters DNA methylation of placental leptin and adiponectin genes in humans, as well as thousands of other genes in umbilical cord tissue and blood that have been implicated in obesity.38
A complete review of epigenetic mechanisms in the genesis of obesity is beyond the scope of this article and can be accessed in other reports.8,40
CONCLUSION
Genetic predisposition is an essential component in the genesis of obesity. Rare cases of monogenic and syndromic obesity have a well-established genetic background, and this knowledge has contributed to revealing important molecular mechanisms in the pathophysiology of obesity. The diagnosis of these forms of obesity is important because it allows genetic counseling and, in some cases, guides the treatment in a more specific way, as in Prader Willi Syndrome and leptin/melanocortin pathway mutations.
The polygenic nature of common obesity makes the discovery of risk genes and their variants a challenging task. GWAS studies have brought new insights to the understanding of the genesis of obesity. However, the contribution of specific genes to the phenotype of polygenic obesity still accounts for only a small part of BMI variability. Recently the development of GPS has proven to be a valid risk score to identify individuals at higher risk for developing obesity, but still without a place in clinical practice.
It is hoped that in the future, greater knowledge of the contribution of genetic and epigenetic variants to the genesis of obesity will assist physicians in clinical decision making, as early and intense preventive measures for people with a high genetic risk score for the development of obesity, and personalized treatment of the obese based on the genetic background.
CONFLICTS OF INTEREST
The authors declare that they do not have any conflicts of interest.


