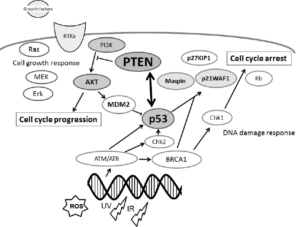
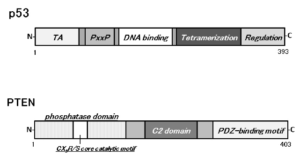
1. Perry ME and Levine AJ. Tumor-suppressor p53 and the cell cycle. Curr Opin Genet Dev. 1993; 3(1): 50-54.doi: 10.1016/s0959-437x(05)80340-5
2. Wang B, Xiao Z, Ko HL, and Ren EC. The p53 response element and transcriptional repression. Cell Cycle. 2010; 9(5): 870- 879. doi: 10.4161/cc.9.5.10825
3. Murray-Zmijewski F, Slee EA, and Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008; 9(9): 702-712. doi: 10.1038/nrm2451
4. Song MS, Salmena L, and Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012; 13(5): 283-296. doi: 10.1038/nrm3330
5. Merritt MA and Cramer DW. Molecular pathogenesis of endometrial and ovarian cancer. Cancer Biomark. 2010; 9(1-6): 287-305. doi: 10.3233/CBM-2011-0167
6. Hobert JA and Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009; 11(10): 687-694. doi: 10.1097/GIM.0b013e3181ac9aea
7. Maehama T and Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999; 9(4): 125-128. doi: http://dx.doi.org/10.1016/S0962- 8924(99)01519-6
8. Chen Z, Trotman LC, Shaffer D et al. Crucial role of p53- dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005; 436(7051): 725-730. doi: 10.1038/ nature03918
9. Kim J, Eltoum IE, Roh M, Wang J, and Abdulkadir SA. Interactions between cells with distinct mutations in c-MYC and Pten in prostate cancer. PLoS Genet. 2009; 5(7): e1000542. doi: 10.1371/journal.pgen.1000542
10. Blanco-Aparicio C, Renner O, Leal JF, and Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007; 28(7): 1379-1386. doi: 10.1093/carcin/bgm052
11. Faesen AC, Dirac AM, Shanmugham A, Ovaa H, Perrakis A, and Sixma TK. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol Cell. 2011; 44(1): 147-159. doi: 10.1016/j.molcel.2011.06.034
12. Sacco JJ, Coulson JM, Clague MJ, and Urbé S. Emerg ing roles of deubiquitinases in cancer-associated pathways. IUBMB Life. 2010; 62(2): 140-157. doi: 10.1002/iub.300
13. Cully M, You H, Levine AJ, and MAKTW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesism. Nat Rev Cancer. 2006; 6(3): 184-192. doi: 10.1038/nrc1819
14. Okumura N, Yoshida H, Kitagishi Y, Murakami M, Nishimura Y, and Matsuda S. PI3K/AKT/PTEN Signaling as a Molecular Target in Leukemia Angiogenesis. Adv Hematol. 2012; 2012: 843085. doi: http://dx.doi.org/10.1155/2012/843085
15. Okumura N, Yoshida H, Kitagishi Y, Nishimura Y, and Matsuda S. Alternative splicings on p53, BRCA1 and PTEN genes involved in breast cancer. Biochem Biophys Res Commun. 2011; 413(3): 395-399. doi: 10.1016/j.bbrc.2011.08.098
16. Walden H and Martinez-Torres RJ. Regulation of Parkin E3 ubiquitin ligase activity. Cell Mol Life Sci. 2012; 69(18): 3053- 3067. doi: 10.1007/s00018-012-0978-5
17. Mueller S, Phillips J, Onar-Thomas A et al. PTEN promoter methylation and activation of the PI3K/AKT/mTOR pathway in pediatric gliomas and influence on clinical outcome. Neuro Oncol. 2012; 14(9): 1146-1152. doi: 10.1093/neuonc/nos140
18. Faurschou A, Gniadecki R, Calay D, and Wulf HC. TNFalpha impairs the S-G2/M cell cycle checkpoint and cyclobutane pyrimidine dimer repair in premalignant skin cells: role of the PI3K-AKT pathway. J Invest Dermatol. 2008; 128(8): 2069- 2077. doi: 10.1038/jid.2008.19
19. Chen Y, Wang BC, and Xiao Y. PI3K: a potential therapeutic target for cancer. J Cell Physiol. 2012; 227(7): 2818-2821.doi: 10.1002/jcp.23038
20. Choi Y, Zhang J, Murga C et al. PTEN, but not SHIP and SHIP2, suppresses the PI3K/AKT pathway and induces growth inhibition and apoptosis of myeloma cells. Oncogene. 2002; 21(34): 5289-5300. doi: 10.1038/sj.onc.1205650
21. Petrella BL, and Brinckerhoff CE. PTEN suppression of YY1 induces HIF-2 activity in von-Hippel-Lindau-null renalcell carcinoma. Cancer Biol Ther. 2009; 8(14): 1389-1401. doi: 10.4161/cbt.8.14.8880
22. Yamamoto M, Tamakawa S, Yoshie M, Yaginuma Y, and Ogawa K. Neoplastic hepatocyte growth associated with cy clin D1 redistribution from the cytoplasm to the nucleus in mouse hepatocarcinogenesis. Mol Carcinog. 2006; 45(12): 901-913. doi: 10.1002/mc.20204
23. Andrés-Pons A, Gil A, Oliver MD, Sotelo NS, and Pulido R. Cytoplasmic p27Kip1 counteracts the pro-apoptotic function of the open conformation of PTEN by retention and destabilization of PTEN outside of the nucleus. Cell Signal. 2012; 24(2): 577- 587. doi: 10.1016/j.cellsig.2011.10.012
24. Planchon SM, Waite KA, and Eng C. The nuclear affairs of PTEN. J Cell Sci. 2008; 121(Pt 3): 249-253. doi: 10.1242/ jcs.022459
25. Pei D, Zhang Y, and Zheng J. Regulation of p53: a collaboration between MDM2 and Mdmx. Oncotarget. 2012; 3(3): 228- 235.doi: 10.18632/oncotarget.443
26. Palmero EI, Achatz MI, Ashton-Prolla P, Olivier M, and Hainaut P. Tumor protein 53 mutations and inherited cancer: beyond Li-Fraumeni syndrome. Curr Opin Oncol. 2010; 22(1): 64-69. doi: 10.1097/CCO.0b013e328333bf00
27. Vousden KH and Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009; 137: 413–431. doi: 10.1016/j.cell.2009.04.037
28. Kruse JP and Gu W. Modes of p53 regulation. Cell. 2009; 137: 609–622. doi: 10.1016/j.cell.2009.04.050
29. Freeman DJ, Li AG, Wei G et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatasedependent and -independent mechanisms. Cancer Cell. 2003; 3(2): 117-130.
30. Lo PK, Lee JS, and Sukumar S. The p53-p21WAF1 checkpoint pathway plays a protective role in preventing DNA rereplication induced by abrogation of FOXF1 function. Cell Signal. 2012; 24(1): 316-324. doi: 10.1016/j.cellsig.2011.09.017
31. Puszyński K, Hat B, and Lipniacki T. Oscillations and bistability in the stochastic model of p53 regulation. J Theor Biol. 2008; 254(2): 452-465. doi: 10.1016/j.jtbi.2008.05.039
32. Eitel JA, Bijangi-Vishehsaraei K, Saadatzadeh MR et al. EN and p53 are required for hypoxia induced expression of maspin in glioblastoma cells. Cell Cycle. 2009; 8(6): 896-901. doi: 10.4161/cc.8.6.7899
33. Zhang M. PTEN in action: coordinating with p53 to regulate maspin gene expression. Cell Cycle. 2009; 8(8): 1112-1113. doi: 10.4161/cc.8.8.8506
34. Mayo LD and Donner DB. A phosphatidylinositol 3-kinase/ AKT pathway promotes translocation of MDM2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001; 98(20): 11598-11603.doi: 10.1073/pnas.181181198
35. Quevedo C, Kaplan DR, and Derry WB. AKT-1 regulates DNA-damage-induced germline apoptosis in C. elegans. Curr Biol. 2007; 17(3): 286-292. doi: http://dx.doi.org/10.1016/j. cub.2006.12.038
36. Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, and Hung MC. HER-2/neu induces p53 ubiquitination via AKT-mediated MDM2 phosphorylation. Nat Cell Biol. 2001; 3(11): 973-982. doi: 10.1038/ncb1101-973
37. Ming M and He YY. PTEN in DNA damage repair. Cancer Lett. 2012; 319(2): 125-129. doi: 10.1016/j.canlet.2012.01.003
38. Bonavida B and Baritaki S. The novel role of Yin Yang 1 in the regulation of epithelial to mesenchymal transition in cancer via the dysregulated NF-κB/Snail/YY1/RKIP/PTEN Circuitry. Crit Rev Oncog. 2011; 16(3-4): 211-226. doi: 10.1615/CritRevOncog.v16.i3-4.50
39. Wang X and Jiang X. MDM2 and MdmX partner to regulate p53. FEBS Lett. 2012; 586(10): 1390-1396. doi: 10.1016/j.febslet.2012.02.049
40. Levav-Cohen Y, Haupt S, and Haupt Y. MDM2 in growth signaling and cancer. Growth Factors. 2005; 23(3): 183-192. doi: 10.1080/08977190500196218
41. Mayo LD and Donner DB. The PTEN, MDM2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002; 27(9): 462-467. doi: http://dx.doi.org/10.1016/S0968-0004- (02)02166-7
42. Kirch HC, Flaswinkel S, Rumpf H, Brockmann D, and Esche H. Expression of human p53 requires synergistic activation of transcription from the p53 promoter by AP-1, NF-kappaB and Myc/Max. Oncogene. 1999; 18(17): 2728-2738. doi: 10.1038/sj.onc.1202626
43. Zheng T, Meng X, Wang J et al. PTEN- and p53-mediated apoptosis and cell cycle arrest by FTY720 in gastric cancer cells and nude mice. J Cell Biochem. 2010; 111(1): 218-228. doi: 10.1002/jcb.22691
44. Mayo LD, Dixon JE, Durden DL, Tonks NK, and Donner DB. PTEN protects p53 from MDM2 and sensitizes cancer cells to chemotherapy. J Biol Chem. 2002; 277(7): 5484-5489. doi: 10.1074/jbc.M108302200
45. Sen P, Mukherjee S, Ray D, and Raha S. Involvement of the AKT/PKB signaling pathway with disease processes. Mol Cell Biochem. 2003; 253(1-2): 241-246. doi: 10.1023/A:1026020101379
46. de Lima Mde D, Marques YM, Alves Sde M Jr et al. MDM2, P53, P21WAF1 and pAKT protein levels in genesis and behaviour of adenoid cystic carcinoma. Cancer Epidemiol. 2009; 33(2): 142-146. doi: 10.1016/j.canep.2009.04.016
47. Li AG, Piluso LG, Cai X, Wei G, Sellers WR, and Liu X. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol Cell. 2006; 23(4): 575-587. doi: http://dx.doi.org/10.1016/j.molcel.2006.06.028
48. Vu BT and Vassilev L. Small-molecule inhibitors of the p53- MDM2 interaction. Curr Top Microbiol Immunol. 2011; 348: 151-172. doi: 10.1007/82_2010_110
49. Bixby D, Kujawski L, Wang S, and Malek SN. The pre-cli nical development of MDM2 inhibitors in chronic lymphocytic leukemia uncovers a central role for p53 status in sensitivity to MDM2 inhibitor-mediated apoptosis. Cell Cycle. 2008; 7(8): 971-979. doi: 10.4161/cc.7.8.5754
50. Amodio N, Scrima M, Palaia L et al. Oncogenic role of the E3 ubiquitin ligase NEDD4-1, a PTEN negative regulator, in non-small-cell lung carcinomas. Am J Pathol. 2010; 177(5): 2622-2634. doi: 10.2353/ajpath.2010.091075
51. Valiente M, Andrés-Pons A, Gomar B et al. Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/ threonine kinases. J Biol Chem. 2005; 280(32): 28936-28943. doi: 10.1074/jbc.M504761200
52. Torres J and Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001; 276(2): 993-998. doi: 10.1074/jbc. M009134200
53. Wu W, Wang X, Zhang W et al. Zinc-induced PTEN protein degradation through the proteasome pathway in human airway epithelial cells. J Biol Chem. 2003; 278(30): 28258-28263. doi: 10.1074/jbc.M303318200
54. Li Y, Ozaki T, Kikuchi H, Yamamoto H, Ohira M, and Nakagawara A. A novel HECT-type E3 ubiquitin protein ligase NEDL1 enhances the p53-mediated apoptotic cell death in its catalytic activity-independent manner. Oncogene. 2008; 27(26): 3700-3709. doi: 10.1038/sj.onc.1211032
55. ShinadAKTsukiyama T, Sho T, Okumura F, Asaka M, and Hatakeyama S. RNF43 interacts with NEDL1 and regulates p53- mediated transcription. Biochem Biophys Res Commun. 2011; 404(1): 143-147. doi: 10.1016/j.bbrc.2010.11.082
56. Barata JT. The impact of PTEN regulation by CK2 on PI3K-dependent signaling and leukemia cell survival. Adv Enzyme Regul. 2011; 51(1): 37-49. doi: 10.1016/j.advenzreg.2010.09.012
57. Kang JY, Kim JJ, Jang SY, and Bae YS. The p53-p21(Cip1/WAF1) pathway is necessary for cellular senescence induced by the inhibition of protein kinase CKII in human colon cancer cells. Mol Cells. 2009; 28(5): 489-494. doi: 10.1007/s10059-009-0141-9
58. Vousden KH and Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007; 8(4): 275-283. doi: 10.1038/nrm2147
59. Weinberg RL, Veprintsev DB, Bycroft M, and Fersht AR. Comparative binding of p53 to its promoter and DNA recognition elements.J Mol Biol. 2005; 348(3): 589-596. doi: 10.1016/j.jmb.2005.03.014
60. Lahav G. The strength of indecisiveness: oscillatory behavior for better cell fate determination. Sci STKE. 2004; 2004(264): 55. doi: 10.1126/stke.2642004pe55
61. Zhang T, Brazhnik P, and Tyson JJ. Exploring mechanisms of the DNA-damage response: p53 pulses and their possible relevance to apoptosis. Cell Cycle. 2007; 6(1); 85-94. doi: 10.4161/ cc.6.1.3705
62. Zhang XP, Liu F, Cheng Z, and Wang W. Cell fate decision mediated by p53 pulses. Proc Natl Acad Sci U S A. 2009; 106(30): 12245-12250. doi: 10.1073/pnas.0813088106
63. Rahal OM and Simmen RC. PTEN and p53 cross-regulation induced by soy isoflavone genistein promotes mammary epithelial cell cycle arrest and lobuloalveolar differentiation. Carcinogenesis. 2010; 31(8): 1491-1500. doi: 10.1093/carcin/bgq123
64. Zhang X, Chen X, Zhai Y, Cui Y, Cao P, Zhang H, Wu Z, Li P, Yu L, Xia X, He F, and Zhou G. Combined Effects of Genetic Variants of the PTEN, AKT1, MDM2 and p53 Genes on the Risk of Nasopharyngeal Carcinoma. PLoS One. 2014; 9(3): e92135. doi: 10.1371/journal.pone.0092135
65. Han CT, Schoene NW, and Lei KY. Influence of zinc deficiency on Akt-Mdm2-p53 and Akt-p21 signaling axes in normal and malignant human prostate cells. Am J Physiol Cell Physiol. 2009; 297(5): C1188-1199. doi: 10.1152/ajpcell.00042.2009