INTRODUCTION
The severity of brain injury is the main determinant of morbidity and mortality in neurocritical patients. However, the role of other associated extracranial complications should not be disregarded1 with pulmonary complications being among the most common ones (Figure 1).2 Major respiratory complications associated with injury to the Central Nervous System (CNS) are caused by airways dysfunction (inability to maintain passage due to neurologic depression or damage), respiratory muscles dysfunction (nerve injury) or intrinsic pulmonary disorders (infection, embolism, acute respiratory distress syndrome, etc.). The occurrence of such complications could potentially cause hypoxemia, which would secondarily aggravate the brain damage. This situation is known to account for up to 50% deaths resulting from brain injury and is considered to be an independent factor for mortality.1,3,4,5,6,7,8
Figure 1: Extracerebral complications in neurocritics patient.
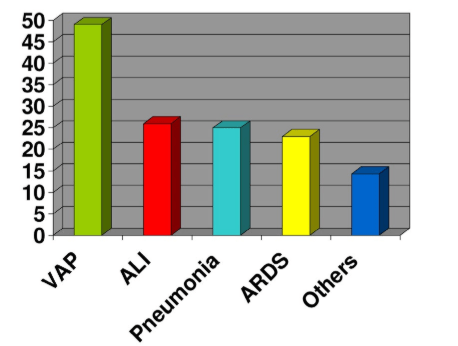
VAP: Ventilator-Associated Pneumonia; ALI: Acute Lung Injury; ARSD: Adult Respiratory Distress Syndrome
The objective of the present article was to offer an up to date review of theories and available evidence on relation between the cerebral alterations and the interactions to pulmonary level.
PATHOPHYSIOLOGY
The interaction between the CNS and the respiratory system is evidenced at the physiological level since the respiratory center, located in the brain stem, controls involuntary respiratory activity and a second, less defined, center in the brain controls for the voluntary respiratory activity.
The ventilatory activity of airways and chest muscles is triggered by spinal cord and carotid sinuses, which are in turn stimulated by CO2 and Hydrogen ion [H+] concentrations. Carotid sinuses also respond to changes in PaO2. Current theories proposed to explain the pathophysiological mechanisms of respiratory complications in case of brain injury are summarized as follows (Figure 2).
Figure 2: Pathophysiology of lung complications in patients with brain injury.
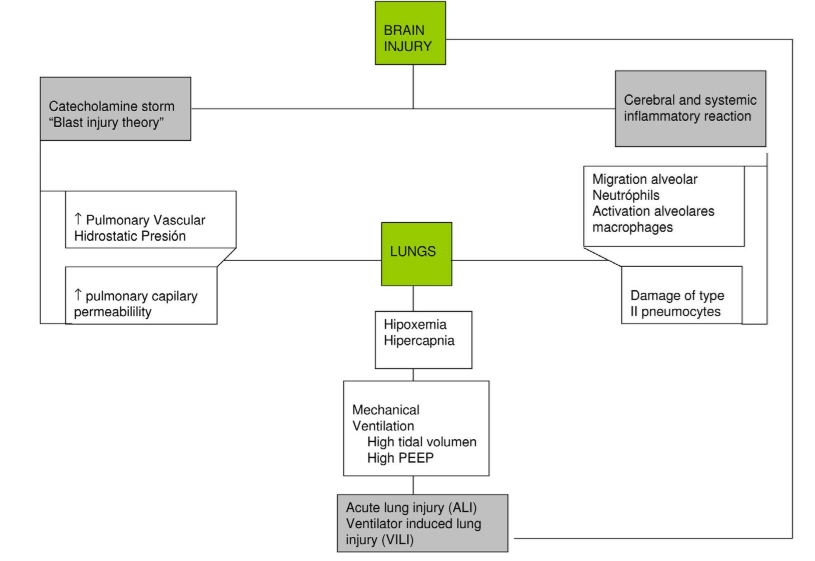
Sympathetic Storm
When brain injury occurs, an initial sympathetic discharge elevates plasmatic adrenaline levels to about 1200 times the normal value within seconds. Although the level of adrenaline subsequently falls, it keeps 3 times higher than normal for about ten days.2,3,7,9,10 This catecholamine release elevates intravascular pressure thus damaging the endothelium and producing pulmonary edema due to disruption of the alveolar-capillary barrier. The initial hydrostatic edema becomes a protein-rich edema that goes into interstitial and alveolar spaces.3,7,9,10 The amount of fluid that leaks through the endothelium depends on the severity of capillary hypertension. Furthermore, in case of structural damage to the capillary wall, plasma will pass to the interstitial and alveolar spaces. The medulla oblongata is considered to be the key anatomical structure directly involved in this pathophysiological response; experiments demonstrated that bilateral lesions to this structure cause systemic hypertension and lung injury.11 Additionally, the possible involvement of the hypothalamus (dysfunction of the vasomotor center), elevated Intracranial pressure (ICP) or activation of the sympathetic-adrenal system have also been studied.11
Inflammatory Theory
As part of the inflammatory response to acute brain injury, intracranial production of cytokines may increase and the permeability of the blood-brain barrier may be higher. The resulting transcranial release of pro-inflammatory cytokines could cause migration of neutrophils and activation of macrophages in the alveolar spaces, as well as structural damage to type II pneumocytes, thus producing secondary lung damage.12,13,14,15,16,17,18
Double Injury Theory (Double Hit Model)
Brain injury would produce a systemic inflammatory response, which would increase vulnerability to inflammatory processes well tolerated in normal situations, such as infection, mechanical stress from artificial ventilation or surgical procedures. Such inflammatory processes could in turn worsen the initial brain damage, harm distant organs and cause multiorgan dysfunction.19,20,21,22,23
After severe brain injury, respiratory failure is the commonest associated dysfunction. In such cases, pathophysiological processes to be considered are: preclinical lung injury, altered capillary permeability with activated neutrophils and macrophages migrating into the airways and alveolar spaces, increased concentrations of pro-inflammatory cytokines in bronchoalveolar lavage and damage to type II pneumocytes – characterized by intense cell vacuolation and lipid peroxidation of lung tissue that causes lysis of the cell membranes. All of this would make lungs more susceptible to secondary damage (“double lesion model”) that could result from initial trauma, respiratory strategy, infection, transfusion, etc.19,20,21,22,23
These experimental and clinical data support the hypothesis that after severe brain injury eventually leading to brain death, preclinical lung injury occurs. The catecholamine storm and the systemic production of inflammatory mediators create a systemic inflammatory environment where the lung is more susceptible to further injurious stimuli, such as ventilatory settings, infections, and transfusions.
CLINICAL PICTURE
Neurogenic Pulmonary Edema (NPE)
Neurogenic Pulmonary Edema is a complication of severe CNS injury that develops early after trauma in the absence of prior cardiac or pulmonary dysfunction. It was first described in 1908 by Shanahan,24 who reported the finding of pulmonary edema in 11 patients with epilepsy. It is occasionally classified as a type of acute respiratory distress, although its pathophysiology and prognosis are different (Table 1). Its severity is related to the magnitude of the brain injury and it is associated with high morbidity rates and up to 7% mortality.8,10,25 NPE may be triggered by different brain conditions, including seizures, stroke and traumatic brain injury (Figure 3). NPE is most common in seizures during the post-critical period, affecting to up to one third of patients in status epilepticus.26,27
| Table 1: Pathophysiology types of lung edema. |
|
↑ Hydrostatic
Pressure |
↑ Permeability |
| Cardiogenic pulmonary
edema |
++++ |
|
|
Reexpansion pulmonary
edema
|
++++ |
++ |
| Neonatal respiratory
distress syndrome |
++ |
++++ |
|
Neurogenic pulmonary
edema
|
++++ |
++++ |
| High altitude pulmonary
edema |
++++ |
++ |
|
Adult respiratory distress
syndrome / Acute lung injury
|
|
++++ |
| Negative pressure
pulmonary edema |
++++ |
++++ |
Figure 3: Neurogenic pulmonary edema associated with intracelebral homorrhage.
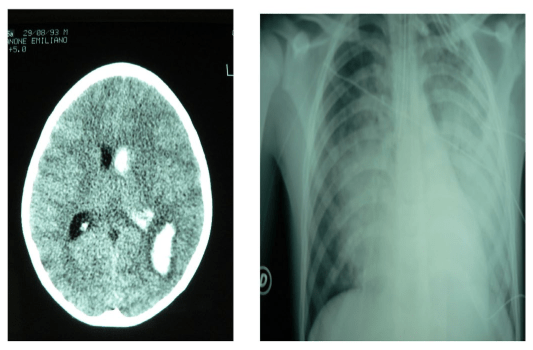
Among patients with brain hemorrhage, NPE mostly occurs in those with Subarachnoid hemorrhage (SAH) from ruptured aneurysms and is correlated with the clinical and radiological severity of bleeding.24,25,26
In head injury the development of pulmonary edema associated with intracranial hypertension %), although not necessary, with a lower incidence (around 1%).26 Clinical presentation of NPE typically comprises early onset and rapid development during the first few hours or days after brain injury; it usually resolves within 24-48 hours. Its most common symptom is dyspnea, accompanied by tachypnea, tachycardia, and crackles at baseline. It should be diagnosed in the presence of pulmonary edema and after excluding other possible causes. NPE diagnosis is different from that of other types of pulmonary edema (Table 1).
Changes in the Ventilation/Perfusion Ratio
Described in neurocritical patients with hypoxemia in the absence of pulmonary infiltrate (clinical-X ray dissociation), probably related to an alteration in the ventilation/perfusion ratio. It is caused by redistribution of regional perfusion, pulmonary microembolism with increased dead space and removal of pulmonary surfactants due to excessive sympathetic stimulation and hyperventilation.
Acute Lung Injury/Acute Respiratory Distress Syndrome
The occurrence and severity of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS) in patients with severe brain damage mainly depend on: the systemic inflammatory response associated with the release of mediators (cytokines, interleukins, tissue factors, etc.),14,15,16,17,18 the massive release of catecholamines into the bloodstream resulting from brain damage and the resuscitation maneuvers that have been used.3,10 All of this increases lung capillary permeability and results in bilateral pulmonary infiltrates that lead to severe hypoxemia with a low PaO2/FiO2 ratio (<300 mm Hg in ALI, <200 mm Hg in ARDS) and decreased lung compliance.
ALI/ARDS occurs in between 20-25% of patients with severe brain damage and constitutes an independent risk factor that worsens prognosis and increases mortality in these patients. Although direct correlation with the CT-scan images of lesions has not been demonstrated, evidence indicates that patients with the larger, more severe brain injury (larger evacuated mass or displacements from midline) are at 5 to 10 times higher risk of developing ALI.3,7
ALI/ARDS develops relatively soon with an initial peak 2-3 days after starting mechanical ventilation or late at the week.
Risk factors for developing ALI/ARDS include: structural damage observable in initial brain CT-scan study, early detection of low Glasgow Coma Score (GCS), use of vasoactive drugs and history of drug addiction.3,7,28
Pneumonia Associated with Mechanical Ventilation
Mechanical Ventilation-Associated Pneumonia (VAP) is defined as pneumonia developed 48-72 hours after intubation. Many patients with severe brain damage require artificial respiratory support, often prolonged, with the consequent risk of lung infection and prolonged stay in the Intensive Care Unit. The incidence of VAP is 9-27% of all intubated patients and increases with the duration of mechanical ventilation. It is even higher in neurocritical patients, where variable incidence rates oscillate between 30% and 50%.29 VAP mostly appears during the first 4 days. The most commonly involved pathogens are: Pseudomona aeruginosa, Escherichia coli, Klebsiella pneumoniae and Acinetobacter, although in patients with head trauma Staphylococcus aureus is the most common one.30 Independent risk factors for the development of VAP are: low level of consciousness, aspiration, emergency intubation, mechanical ventilation for more than 3 days, reintubation, age over 60 years, supine position, comorbidities and previous antibiotic therapy.30
Mortality rates attributed to VAP are about 30%; this condition doubles the risk of death. It is also estimated to increases the duration of stay at the ICU in 6 days and the associated expenses in 10,000 dollars.30
Mechanical ventilation is the main supportive therapy used to re-establish sufficient oxygen supply and remove Carbon dioxide (CO2) produced by peripheral organs during acute respiratory failure in patients with severe brain injury. While tight CO2 control represents a priority in patients with severe brain injury, optimal treatment of ALI/ARDS consists of a protective ventilatory strategy allowing a certain degree of hypercapnia to protect the lung from further injurious stimuli while it recovers from the initial pathological process.
All these findings support the hypothesis that, after severe brain injury eventually leading to brain death, a preclinical lung injury characterized by an inflammatory response is present. Massive brain injury may act as a preconditioning factor rendering the lung more susceptible to subsequent lung damage induced by mechanical ventilation.
PREVENTION AND TREATMENT OF PULMONARY COMPLICATIONS
Associated with Brain Injury
Management of Neurogenic Pulmonary Edema (NPE)
Neurocritical patients with impaired respiratory function or signs of low cardiac output require strict hemodynamic and echocardiographic monitoring. These patients may potentially need a pulmonary artery catheter (Swan-Ganz). Fluid restriction as normal filling pressure is reached and use beta blockers to reduce oxygen consumption (VO2) may be necessary to improve lung and brain function. Vasopressor agents should be used rationally to avoid possible vasoconstriction of pulmonary vessels or worsening of lung injury due to hemodynamic stress. Respiratory support with supplemental oxygen should be provided with the aim of maintaining SaO2> 94%. Mechanical ventilation should be indicated if necessary.27 The respiratory strategy should include alveolar recruitment and protection in order to avoid ventilation-induced injury (see below).
Prevention of Collapse /Atelectasis in Decline Areas
Mechanical ventilation with tidal volumes of 6 ml/kg, plateau pressure less than 30 cm H2O and PEEP less than 15 cm H2O ensures homogenization of pulmonary ventilation preventing lung collapse that avoid hemodynamic variations which impact on Cerebral Perfusion Pressure (CPP). It has been shown that use of high tidal volumes (9-11 ml/kg) stimulates inflammatory response and lung injury induced by the ventilator.10,25,31,32
Even neurocritical patient’s ventilation in prone possition results in improved brain tissue oxygenation in severe hypoxemia with minimal effect on Intracranial pressure (ICP) and Cerebral Perfusion Pressure (CPP).25,33,34,35,36
Prevention of Mechanical Ventilation-Associated
Pneumonia
Prolonged respiratory support should be avoided; mechanical ventilation should be discontinued as soon as possible.32,34However, in cases of neurocritical disease prompted, patients that require high doses of sedatives or failure to protect the airways (alteration of respiratory center, damage to the nuclei of cranial nerves or ARDS), a tracheostomy may improve patient’s comfort and management of tracheobronchial secretions (Table 2), as well as early onset of chest physiotherapy.36
Table 2: Indications for tracheostomy in neurocritic patients.
- Neurologic stability
- Normal Intracraneal pressure
- Weaning posibility
- Pronlonged alteration of consciousness
- Imposibility to protect airways
- Easier management of pulmonary secretions
- Obstruction of airways
Prophylactic antibiotic therapy against multiresistant pathogens is usually started until the microbiology laboratory results are available. The treatment usually lasts for 7 days if the patient progresses favourably; although it might be extended in patients where Pseudomonas aeruginosa is identified since infection by this organism is associated with high rates of relapse.37
CONCLUSIONS
In patients with severe brain damage, respiratory complications are frequent. Increasing our knowledge of the pathophysiologic mechanisms triggering this situation is essential to enhance prevention and treatment. Key elements to be highlighted include worsening of brain injury secondary to hypoxia and lung damage, and aggravation of lung injury due to inadequate respiratory strategy.


