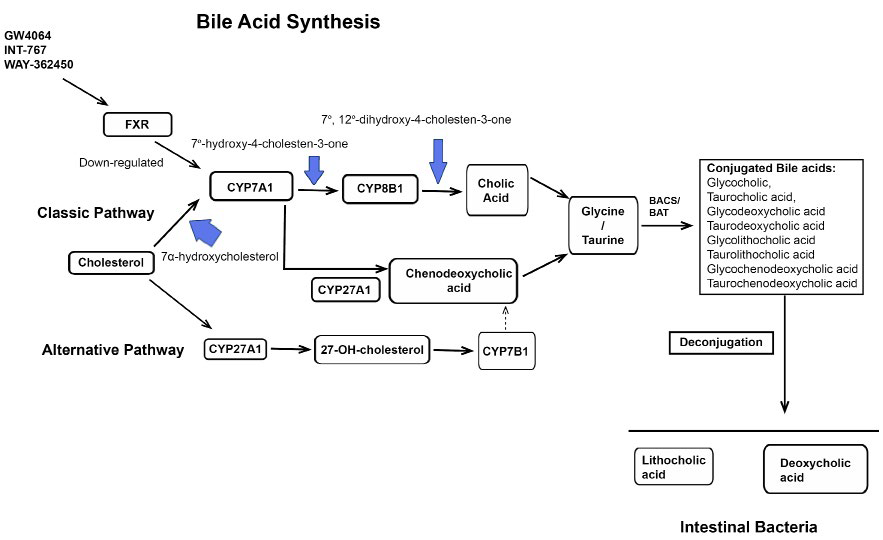
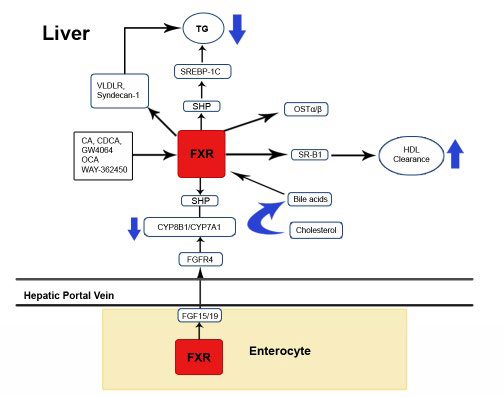
1. Nie B, Park MH, Kazantzis M, et al. Specific bile acids inhibit hepatic fatty acid uptake in mice. Hepatology. 2012; 56: 1300-1310.
doi: 10.1002/hep.25797
2. Li T, Chiang LYJ. Nuclear receptors in bile acid metabolism. Drug Metab Rev. 2013; 45: 145-155. doi: 10.3109/03602532.2012.740048
3. Li Y, Jadhav K, Zhang Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochemical Pharmacology. 2013; 86: 1517-1524.
doi: 10.1016/j.bcp.2013.08.015
4. Hylemon BP, Zhou H, Pandak MW, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. Journal of lipid research. 2012; 50: 1509-1520.
doi: 10.1194/jlr.R900007-JLR200
5. Chiang LYJ. Bile acids: regulation of synthesis. Journal of lipid research. 2009; 50: 1955-1966. doi: 10.1194/jlr.R900010-JLR200
6. Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999; 284:1365-1368.
7. Vlahcevic ZR, Heuman DM, Hylemon BP. Physiology and pathophysiology of enterohepatic circulation of bile acids. Hepatology: A Textbook of Liver Disease. In: Zakim D, Boyer TD,eds. WB Sanders Co, Philadelphia, PA, USA. 1996; 376-417.
8. Makishima M, Okamoto YA, Repa JJ, et al. Identification of a nuclear receptor for bile acids. Science. 1999; 284: 1362-1365.
doi: 10.1126/science.284.5418.1362
9. Staudinger JL, Goodwin B, Jones AS, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA. 2001; 98: 3369-3374. doi: 10.1073/pnas.051551698
10. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011; 141: 1572-1585.
doi: 10.1053/j.gastro.2011.09.002
11. Claudel T, Zollner G, Wagner M, Trauner M. Role of nuclear receptors for bile acid metabolism, bile secretion, cholestasis, and gallstone disease. Biochim Biophys Acta. 2011; 1812: 867-878. doi: 10.1016/j.bbadis.2010.12.021
12. Chiang LYJ. Front. Biosci. 1998; 3: 176-193.
13. Mazuy C, Helleboid A, Staels B, Philippe Lefebvre. Nuclear bile acid signaling through the farnesoid X receptor. Cell Mol Life Sci. 2014; 72: 1631-1650. doi: 10.1007/s00018-014-1805-y
14. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003; 72: 137-174.
doi: 10.1146/annurev.biochem.72.121801.161712
15. Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease: Tematic Review Series: New Lipid and Lipoprotein Targets for the Treatment of Cardiometabolic Diseases. J Lipid Res.2012; 53: 1723-1737.
doi: 10.1194/jlr.R024794
16. Russell DW, Setchell KD. Bile acid biosynthesis. Biochemistry. 1992; 31(1): 4737-4749.
17. Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev. 2004; 36: 703-722.
18. Pandak WM , Ren S, Marques D, et al. Transport of cholesterol into mitochondria is rate-limiting for bile acid synthesis via the alternative pathway in primary rat hepatocytes. J Biol Chem. 2002; 277: 48158 -48164. doi: 10.1074/jbc.M205244200
19. Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008; 65: 2461-2483. doi: 10.1007/s00018-008-7568-6
20. Bernstein C, Holubec H, Bhattacharyya KA, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Archives of Toxicology. 2011; 85: 863-871. doi: 10.1007/s00204-011-0648-7
21. Hofmann FA. The Continuing Importance of Bile Acids in Liver and Intestinal Disease. JAMA. 1999; 159: 2647-2658.
doi: 10.1001/archinte.159.22.2647
22. Changbumrung S, Tungtrongchitr R, Migasena P, Chamroenngan S. Serum unconjugated primary and secondary bile acids in patients with cholangiocarcinoma and hepatocellular carcinoma. J Med Assoc Thai. 1990; 73: 81-90.
23. Boyer JL. New concepts of mechanisms of hepatocyte bile formation. Physiol Rev. 1980; 60: 303-326.
24. Ananthanarayanan M, Balasubramanian VN, Makishima M, Mangelsdorf JD, Suchy JF. Human bile salt export pump (BSEP) promoter is transactivated by the farnesoid X receptor/bile acid receptor (FXR/BAR). J Biol Chem. 2001; 27: 28857-28865
25. Childs S, Yeh RL, Georges E, Ling V. Identification of a sister gene to P-glycoprotein. Cancer Res. 1995; 55: 2029-2034.
26. Strautnieks SS, Kagalwalla AF, Tanner MS, et al. Identification of a locus for progressive familial intrahepatic cholestasis PFIC2 on chromosome 2q24. Am J Hum Genet. 1997; 61: 630-633. doi: 10.1086/515501
27. Dawson PA, Hubbert M, Haywood J, et al. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005; 280:6960-6968. doi: 10.1074/jbc.M412752200
28. Song KH, Li T, Owsley E, Strom S, Chiang JYL. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol hydroxylase gene expression. Hepatology. 2009; 49: 297-305. doi: 10.1002/hep.22627
29. Zhang S, Wang J, Liu Q. Farnesoid X receptor agonist WAY362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009; 51:380-388. doi: 10.1016/j.jhep.2009.03.025
30. Lomanaco R, Sunny NE, Bril F, Cusi K. Nonalcoholic fatty liver disease: current issues and novel treatment approaches. Drugs. 2013; 73: 1-14. doi: 10.1007/s40265-012-0004-0
31. McNear S, Harrison, SA. Current status of therapy in nonalcoholic fatty liver disease. Therapeutic Advances in Gastroenterology. 2009; 2: 29-43. doi: 10.1177/1756283X08100327
32. Onis M, Blossner M, Borghi E. Mutation research/fundamental and molecular mechanisms of mutagenesis. American Journal of Clinical Nutrition. 2010; 92: 1257-1264.
33. Durazzo M, Belci P, Collo A, Grisoglio E, Bo S. Focus on therapeutic strategies of nonalcoholic fatty liver disease. International Journal of Hepatology. 2012; 1: 1-9. doi: 10.1155/2012/464706
34. Day CP, James OF. Steatohepatitis: a tale of two “hits”?Gastroenterology. 1998; 114: 842-845.
doi: 10.1016/S0016-5085-(98)70599-2
35. Marra F, Gastaldelli AS, Baroni SG, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends in molecular medicine. 2008; 14: 72-81. doi: 10.1016/j.molmed.2007.12.003
36. Adorini L, Pruzanski M, Shapiro D. Farnesoid X receptor targeting to treat non-alcoholic steatohepatitis. Drug Discov Today. 2012; 17: 988-997. doi: 10.1016/j.drudis.2012.05.012
37. Kodama Y, Brenner DA. c-Jun N-Terminal kinase signaling in the pathogenesis of nonalcoholic fatty liver disease: multiple roles in multiple steps. Hepatology. 2008; 49: 6-8. doi: 10.1002/hep.22710
38. Gyamfi D, Everitt H, Tewfik I, Clemens D, Patel V. Hepatic mitochondrial dysfunction induced by fatty acids and ethanol. Free Radical Biology and Medicine. 2012; 53: 2131-2145. doi: 10.1016/j.freeradbiomed.2012.09.024
39. Ratziu V, Pienar L. Pharmacological therapy for non-alcoholic steatohepatitis: How efficient are thiazolidinediones? Hepatology Research. 2011; 41: 687-695. doi: 10.1111/j.1872-034-X.2011.00825.x
40. Aithal GP, Thomas JA, Kaye PV, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008; 135:1176-1184. doi: 10.1053/j.gastro.2008.06.047
41. Sharma AM, Staels B. Peroxisome proliferator-activated receptor gamma and adipose tissue-understanding obesity-related changes in regulation of lipid and glucose metabolism. J Clin Endocrinol Metab. 2007; 92: 386-395. doi: 10.1210/jc.2006-1268
42. Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006; 355: 2297-2307. doi: 10.1056/NEJMoa060326
43. Boettcher E, Csako G, Pucino F, Wesley R, Loomba R. Meta-nalysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2012; 35: 66-75. doi: 10.1111/j.1365-2036.2011.04912.x
44. Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. The New England Journal of Medicine. 2010; 362: 1675-1685. doi:10.1056/NEJMoa0907929
45. Baran B, Akyuz F. Non-alcoholic fatty liver disease: What has changed in the treatment since the beginning? World J Gastroenterol. 2014; 20: 14219-14229. doi: 10.3748/wjg.v20.i39.14219
46. Lavine JE, Schwimmer JB, Van ML. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the tonic randomized controlled trial. JAMA. 2011; 305: 1659-1668. doi: 10.1001/jama.2011.520
47. Saremi A, Arora R. Vitamin E and cardiovascular disease. Am J Ther. 2010; 17: 56-65. doi: 10.1097/MJT.0b013e31819cdc9a
48. Yang ZX, Shen W, Sun H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with nonalcoholic fatty liver disease. Hepatol Int. 2010; 4: 741-748. doi: 10.1007/s12072-010-9202-6
49. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000; 102: 731-744. doi: 10.1016/S0092-8674(00)00062-3
50. Abu-Shanab A, Quigley EM..The role of the gut microbiota in non-alcoholic fatty liver disease. Nat Rev GastroenterolHepatol. 2010; 7: 691-701. doi: 10.1038/nrgastro.2010.172
51. Elsharkawy AM, Mann DA. Nuclear factor-kappaB and thehepatic inflammation-fibrosis-cancer axis. Hepatology. 2007;46: 590-597.
doi: 10.1002/hep.21802
52. Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. 2007; 28: 940-946. doi: 10.1093/carcin/bgl249
53. Carr MR, Reid EA. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm Res. 2015; 17(16): 1-16.
54. Stayrook KR, Bramlett KS, Savkur RS, et al. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. 2005; 146: 984-991.
55. Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J. Bile acids regulate gluconeogenic gene expression via small heterodimer partner mediated repression of hepatocyte nuclear factor 4 and Foxo1. J Biol Chem. 2004; 279: 23158-23165. doi: 10.1074/jbc.M314322200
56. Langhi C, May CL, Kourimate S, et al. Activation of the farnesoid X receptor represses PCSK9 expression in human hepatocytes. FEBS Lett. 2008; 582: 949-955. doi: 10.1016/j.febslet.2008.02.038
57. Ma Y, Huang Y, Yan L, Gao M, Liu D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm Res. 2013; 30: 1447-1457. doi: 10.1007/s11095-013-0986-7
58. Zhang Y, Lee FY, Barrera G, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci, USA. 2006; 103: 1006-1011. doi: 10.1073/pnas.0506982103
59. Cariou B, Harmelen VK, Duran-Sandoval D, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. 2006; 281: 11039-11049. doi:10.1074/jbc.M510258200
60. Xiong X, Wang X, Lu Y, et al. Hepatic steatosis exacerbated by endoplasmic reticulum stress-mediated downregulation of FXR in aging mice. J Hepatol. 2014; 60: 847-854. doi: 10.1016/j.jhep.2013.12.003
61. LiuY, Binz J, Numerick JM, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest. 2003; 112: 1678-1687.doi: 10.1172/JCI200318945
62. Pellicciari R, Fiorucci S, Camaioni E, et al. 6-alpha-ethylchenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002; 45: 3569-3572. doi: 10.1021/jm025529g
63. Mi LZ, Devarakonda S, Harp JM, et al. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol Cell. 2003; 11: 1093-1100. doi: 10.1016/S1097-2765-(03)00112-6
64. Vignozzi L, Morelli A, Filippi S, et al. Farnesoid X receptor activation improves erectile function in animal models of metabolic syndrome and diabetes. Journal of Sexual medicine. 2011;8: 57-77. doi: 10.1111/j.1743-6109.2010.02073.x
65. Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010; 51: 771-784. doi: 10.1194/jlr.M001602
66. Mencarelli A, Renga B, Migliorati M, et al. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. Journal of immunology. 2009; 183: 6657-6666. doi: 10.4049/jimmunol.0901347
67. Fiorucci S, Antonelli E, Rizzo G, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004; 127:1497-1512. doi: 10.1053/j.gastro.2004.08.001
68. Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013; 145: 574-582. doi: 10.1053/j.gastro.2013.05.042
69. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2014; 385: 956-1065. doi: 10.1016/S0140-6736(14)61933-4
70. Walters JR, Johnston IM, Nolan JD, Vassie C, Pruzanski ME, Shapiro DA. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment Pharmacol Ther. 2015; 41: 54-64. doi: 10.1111/apt.12999
71. Ratziu V. Treatment of NASH with ursodeoxycholic acid: Pro. Clin Res Hepatol Gastroenterol. 2012; 36(Suppl 1): S41-S45.
doi: 10.1016/S2210-7401(12)70020-7
72. Xiang Z, Chen YP, Ma KF, et al. The role of ursodeoxycholic acid in non-alcoholic steatohepatitis: a systematic review. BMC Gastroenterol. 2013; 13: 140. doi: 10.1186/1471-230X-13-140
73. Lindor KD, Kowdley KV, Heathcote EJ, et al. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology. 2004; 39: 770-778. doi:10.1002/hep.20092
74. Adams LA, Angulo P, Petz J, Keach J. Lindor KD. A pilot trial of high-dose ursodeoxycholic acid in non-alcoholic Steatohepatitis. Hepatol Int. 2010; 4: 628-633. doi: 10.1007/s12072-010-9195-1
75. Ratziu V1, de Ledinghen V, Oberti F, et al. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011; 54:1011-1019. doi: 10.1016/j.jhep.2010.08.030
76. Cho EJ, Yoon JH, Kwak MS, et al. Tauroursodeoxycholic acid attenuates progression of steatohepatitis in mice fed a methionine-choline-deficient diet. Dig Dis Sci. 2014; 59: 1461-1474. doi: 10.1007/s10620-014-3217-
77. Hafeez S, Ahmed MH. Bariatric surgery as potential treatment for nonalcoholic fatty liver disease: a future treatment by choice or by chance? Journal of Obesity. 2013; Article ID 839275. doi: 10.1155/2013/839275
78. Ben M, Polimeni L, Baratta F, Pastori D, Loffredo L, Angelico F. Modern approach to the clinical management of nonalcoholic fatty liver disease. World J Gastroenterol. 2014; 20:8341-8350. doi:10.3748/wjg.v20.i26.8341
79. Bower G, Toma T, Harling L, et al. Bariatric surgery and non-alcoholic fatty liver disease: a systematic review of liver biochemistry and histology. Obes Surg. 2015. doi: 10.1007/s11695-015-1691-x