1. Downey JM. Free radicals and their involvement during longterm myocardial ischemia and reperfusion. Annu Rev Physiol. 1990; 52: 487-504.
doi: 10.1146/annurev.ph.52.030190.002415
2. Duerr GD, Dewald D, Schmitz EJ, et al. Metallothioneins 1 and 2 modulate inflammation and support remodeling in ischemic cardiomyopathy in mice. Mediators of inflammation. 2016; 2016: 7174127. doi: 10.1155/2016/7174127
3. Lund J, Hafstad AD, Boardman NT, et al. Exercise training promotes cardioprotection through oxygen-sparing action in high fatfed mice. Am J Physiol Heart Circ Physiol. 2015; 308(8): H823-H829. doi: 10.1152/ajpheart.00734.2014
4. Woodall MC, Woodall BP, Gao E, Yuan A, Koch WJ. Cardiac fibroblast GRK2 deletion enhances contractility and remodeling following ischemia/reperfusion injury. Circ Res. 2016; 119(10): 1116-1127. doi: 10.1161/CIRCRESAHA.116.309538
5. Zhou Y, Fang H, Lin S, et al. Qiliqiangxin protects against Cardiac ischemia-reperfusion injury via activation of the mTOR pathway. Cell Physiol Biochem. 2015; 37(2): 454-464. doi: 10.1159/000430368
6. Dauerman HL, Bates ER, Kontos MC, et al. Nationwide analysis of patients with ST-segment-elevation myocardial infarction transferred for primary percutaneous intervention: Findings from the American Heart Association mission: Lifeline program. Circ Cardiovasc Interv. 2015; 8(5).
doi: 10.1161/CIRCINTERVENTIONS.114.002450
7. Harrison RW, Simon D, Miller AL, de Lemos JA, Peterson ED, Wang TY. Association of hospital myocardial infarction volume with adherence to American College of Cardiology/American Heart Association performance measures: Insights from the National Cardiovascular Data Registry. Am Heart J. 2016; 178: 95-101. doi: 10.1016/j.ahj.2016.04.002
8. Boveris A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol. 1984; 105: 429-435. doi: 10.1016/S0076-6879(84)05060-6
9. Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol. 2002; 282(2): C227-C241.
doi: 10.1152/ajpcell.00112.2001
10. McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985; 312(3): 159-163.
doi: 10.1056/NEJM198501173120305
11. Yu BP. Cellular defenses against damage from reactive oxygen species. Physiol Rev. 1994; 74(1): 139-162. doi: 10.1152/physrev.1994.74.1.139
12. Cadenas S. ROS and redox signaling in myocardial ischemiareperfusion injury and cardioprotection. Free Radic Biol Med. 2018; 117: 76-89.
doi: 10.1016/j.freeradbiomed.2018.01.024
13. Groehler At, Kren S, Li Q, et al. Oxidative cross-linking of proteins to DNA following ischemia-reperfusion injury. Free Radic Biol Med. 2018; 120: 89-101 doi: 10.1016/j.freeradbiomed.2018.03.010
14. Tian C, Gao L, Zimmerman MC, Zucker IH. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am J Physiol Heart Circ Physiol. 2018; doi: 10.1152/ajpheart.00602.2017
15. Zweier JL, Fertmann J, Wei G. Nitric oxide and peroxynitrite in postischemic myocardium. Antioxid Redox Signal. 2001; 3(1): 11-22.
doi: 10.1089/152308601750100443
16. Zhang X, Hu H, Luo J, et al. A novel danshensu-tetramethylpyrazine conjugate DT-010 provides cardioprotection through the PGC-1alpha/Nrf2/HO-1 pathway. Biol Pharm Bull. 2017; 40(9): 1490-1498. doi: 10.1248/bpb.b17-00313
17. Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr. 1999; 31(4): 347-366. doi: 10.1023/A:1005427919188
18. Lesnefsky EJ, Gudz TI, Moghaddas S, et al. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol. 2001; 33(1): 37-47. doi: 10.1006/jmcc.2000.1273
19. Hoppel CL, Moghaddas S, Lesnefsky EJ. Interfibrillar cardiac mitochondrial comples III defects in the aging rat heart. Biogerontology. 2002; 3(1-2): 41-44. doi: 10.1023/A:1015251212039
20. Kuksal N, Gardiner D, Qi D, Mailloux RJ. Partial loss of complex I due to NDUFS4 deficiency augments myocardial reperfusion damage by increasing mitochondrial superoxide/hydrogen peroxide production. Biochem Biophys Res Commun. 2018; 498(1): 214-220.
doi: 10.1016/j.bbrc.2018.02.208
21. Moris D, Spartalis M, Tzatzaki E, et al. The role of reactive oxygen species in myocardial redox signaling and regulation. Ann Transl Med. 2017; 5(16): 324. doi: 10.21037/atm.2017.06.17
22. Scandroglio F, Tortora V, Radi R, Castro L. Metabolic control analysis of mitochondrial aconitase: Influence over respiration and mitochondrial superoxide and hydrogen peroxide production. Free Radic Res. 2014; 48(6): 684-693. doi: 10.3109/10715762.2014.900175
23. Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: A possible regulatory mechanism in mitochondrial function. Biochem J. 2001; 358(Pt 1): 147-155. doi: 10.1042/bj3580147
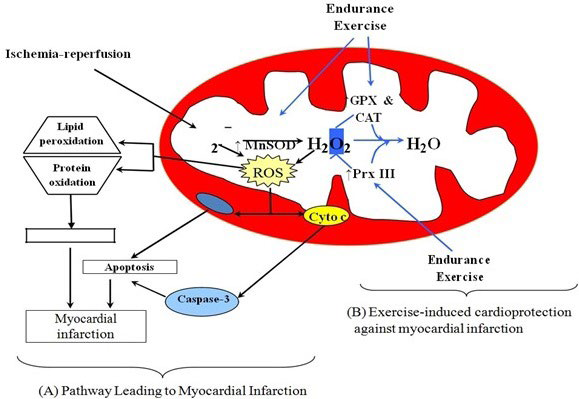
24. Lee Y, Min K, Talbert EE, et al. Exercise protects cardiac mitochondria against ischemia-reperfusion injury. Med Sci Sports Exerc. 2012; 44(3): 397-405. doi: 10.1249/MSS.0b013e318231c037
25. Ribeiro Junior RF, Dabkowski ER, Shekar KC, KA OC, Hecker PA, Murphy MP. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic Biol Med. 2018; 117: 18-29. doi: 10.1016/j.freeradbiomed.2018.01.012
26. Karch J, Molkentin JD. Identifying the components of the elusive mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014; 111(29): 10396-10397. doi: 10.1073/pnas.1410104111
27. Morciano G, Bonora M, Campo G, et al. Mechanistic role of mPTP in ischemia-reperfusion injury. Adv Exp Med Biol. 2017; 982: 169-189.
doi: 10.1007/978-3-319-55330-6_9
28. Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J. 2007; 405(3): 407-415. doi: 10.1042/BJ20070319
29. Lee Y, Gustafsson AB. Role of apoptosis in cardiovascular disease. Apoptosis. 2009; 14(4): 536-548. doi: 10.1007/s10495-008-0302-x
30. Kuo HF, Liu PL, Chong IW, et al. Pigment epithelium-derived factor mediates autophagy and apoptosis in myocardial hypoxia/reoxygenation injury. PLoS One. 2016; 11(5): e0156059. doi: 10.1371/journal.pone.0156059
31. Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest. 1996; 98(5): 1253-1260. doi: 10.1172/JCI118909
32. Van Remmen H, Qi W, Sabia M, et al. Multiple deficiencies in antioxidant enzymes in mice result in a compound increase in sensitivity to oxidative stress. Free Radic Biol Med. 2004; 36(12): 1625-1634. doi: 10.1016/j.freeradbiomed.2004.03.016
33. Van Remmen H, Williams MD, Guo Z, et al. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001; 281(3): H1422-1432. doi: 10.1152/ajpheart.2001.281.3.H1422
34. Chen Z, Siu B, Ho YS, et al. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol. 1998; 30(11): 2281-2289. doi: 10.1006/jmcc.1998.0789
35. Dai DF, Chiao YA, Martin GM, et al. Mitochondrial-targeted catalase: Extended longevity and the roles in various disease models. Prog Mol Biol Transl Sci. 2017; 146: 203-241. doi: 10.1016/bs.pmbts.2016.12.015
36. Izawa S, Inoue Y, Ki mura A. Importance of catalase in the adaptive response to hydrogen peroxide: Analysis of acatalasaemic saccharomyces cerevisiae. Biochem J. 1996; 320 ( Pt 1): 61-67. doi: 10.1042/bj3200061
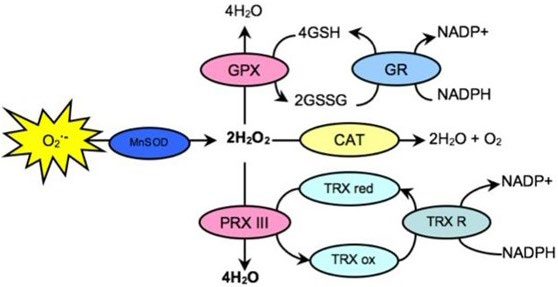
37. Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003; 300(5619): 650-653. doi: 10.1126/science.1080405
38. Rhee SG, Kil IS. Mitochondrial H2O2 signaling is controlled by the concerted action of peroxiredoxin III and sulfiredoxin: Linking mitochondrial function to circadian rhythm. Free Radic Biol Med. 2016; 100: 73-80. doi: 10.1016/j.freeradbiomed.2016.07.029
39. Hofmann B, Hecht HJ, Flohe L. Peroxiredoxins. Biol Chem. 2002; 383(3-4): 347-364. doi: 10.1515/bc.2002.040
40. Kinnula VL, Lehtonen S, Sormunen R, et al. Overexpression of peroxiredoxins I, II, III, V, and VI in malignant mesothelioma. J Pathol. 2002; 196(3): 316-323. doi: 10.1002/path.1042
41. Seaver LC, Imlay JA. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in escherichia coli. J Bacteriol. 2001; 183(24): 7173-7181. doi: 10.1128/JB.183.24.7173-7181.2001
42. Brown DA, Jew KN, Sparagna GC, Musch TI, Moore RL. Exercise training preserves coronary flow and reduces infarct size after ischemia-reperfusion in rat heart. J Appl Physiol. 2003; 95(6): 2510-2518. doi: 10.1152/japplphysiol.00487.2003
43. Hamilton KL, Powers SK, Sugiura T, et al. Short-term exercise training can improve myocardial tolerance to I/R without elevation in heat shock proteins. Am J Physiol Heart Circ Physiol. 2001; 281(3):H1346-H1352. doi: 10.1152/ajpheart.2001.281.3.H1346
44. Lennon SL, Quindry JC, Hamilton KL, et al. Elevated MnSOD is not required for exercise-induced cardioprotection against myocardial stunning. Am J Physiol Heart Circ Physiol. 2004; 287(2):H975-H980. doi: 10.1152/ajpheart.01208.2003
45. Li Y, Cai M, Cao L, et al. Endurance exercise accelerates myocardial tissue oxygenation recovery and reduces ischemia reperfusion injury in mice. PLoS One. 2014; 9(12): e114205. doi: 10.1371/journal.pone.0114205
46. Quindry JC, Hamilton KL, French JP, et al. Exercise-induced HSP-72 elevation and cardioprotection against infarct and apoptosis. J Appl Physiol (1985). 2007; 103(3): 1056-1062. doi: 10.1152/ japplphysiol.00263.2007
47. Hamilton KL, Quindry JC, French JP, et al. MnSOD antisense treatment and exercise-induced protection against arrhythmias. Free Radic Biol Med. 2004; 37(9): 1360-1368. doi: 10.1016/j.freeradbiomed.2004.07.025
48. Yamashita N, Hoshida S, Otsu K, Asahi M, Kuzuya T, Hori M. Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J Exp Med. 1999; 189(11): 1699-1706. doi: 10.1084/jem.189.11.1699
49. Chung YW, Kim HK, Kim IY, Yim MB, Chock PB. Dual function of protein kinase C (PKC) in 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced manganese superoxide dismutase (MnSOD) expression: Activation of CREB and FOXO3a by PKCalpha phosphorylation and by PKC-mediated inactivation of Akt, respectively. J Biol Chem. 2011; 286(34): 29681-29690. doi: 10.1074/jbc.M111.264945
50. Watson PA, Birdsey N, Huggins GS, Svensson E, Heppe D, Knaub L. Cardiac-specific overexpression of dominant-negative CREB leads to increased mortality and mitochondrial dysfunction in female mice. Am J Physiol Heart Circ Physiol. 2010; 299(6): H2056-H2068.
doi: 10.1152/ajpheart.00394.2010
51. Eddy LJ, Goeddel DV, Wong GH. Tumor necrosis factoralpha pretreatment is protective in a rat model of myocardial ischemiareperfusion injury. Biochem Biophys Res Commun. 1992; 184(2): 1056-1059. doi: 10.1016/0006-291X(92)90698-K
52. Pedersen BK. The anti-inflammatory effect of exercise: Its role in diabetes and cardiovascular disease control. Essays Biochem. 2006; 42: 105-117. doi: 10.1042/bse0420105
53. Petersen AM, Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol (1985). 2005; 98(4): 1154-1162.
doi: 10.1152/japplphysiol.00164.2004
54. Smith RE, Tran K, Smith CC, McDonald M, Shejwalkar P, Hara K. The role of the Nrf2/ARE antioxidant system in preventing cardiovascular diseases. Diseases. 2016; 4(4). doi: 10.3390/diseases4040034
55. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008; 103(11): 1232-1240. doi: 10.1161/01.RES.0000338597.71702.ad
56. Shanmugam G, Narasimhan M, Conley RL, et al. Chronic endurance exercise impairs cardiac structure and function in middleaged mice with impaired Nrf2 signaling. Front Physiol. 2017; 8: 268. doi: 10.3389/fphys.2017.00268
57. Gounder SS, Kannan S, Devadoss D, et al. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS One. 2012; 7(9): e45697. doi: 10.1371/journal.pone.0045697
58. Starnes JW, Barnes BD, Olsen ME. Exercise training decreases rat heart mitochondria free radical generation but does not prevent Ca2+-induced dysfunction. J Appl Physiol (1985). 2007; 102(5): 1793-1798. doi: 10.1152/japplphysiol.00849.2006
59. Moran M, Delgado J, Gonzalez B, Manso R, Megias A. Responses of rat myocardial antioxidant defences and heat shockprotein HSP 72 induced by 12 and 24-week treadmill training. Acta Physiol Scand. 2004; 180(2): 157-166. doi: 10.1111/j.0001-6772.2003.01244.x
60. Leeuwenburgh C, Hollander J, Leichtweis S, Griffiths M, Gore M, Ji LL. Adaptations of glutathione antioxidant system to endurance training are tissue and muscle fiber specific. Am J Physiol. 1997; 272(1 Pt 2): R363-R369. doi: 10.1152/ajpregu.1997.272.1.R363
61. Kavazis AN, Alvarez S, Talbert E, Lee Y, Powers SK. Exercise training induces a cardioprotective phenotype and alterations in cardiac subsarcolemmal and intermyofibrillar mitochondrial proteins. Am J Physiol Heart Circ Physiol. 2009; 297(1): H144-H152.
doi: 10.1152/ajpheart.01278.2008