INTRODUCTION
The term ‘Mast Cells’ originated from German term ‘Mastzellen’ that means fattened cells and were named because of large granules in their cytoplasm, which were expected to nourish the surrounding cells. MCs were first characterized by Paul Ehrlich in 1878.1 The MCs originate from myeloid stem cells in bone marrow and the immature cells migrate to different tissues and later on develop into mature cells. MCs are localized in organs rich in connective tissue and are abundantly present in skin, cardiovascular tissues, gastrointestinal tract, and reproductive organs. The immune responses, allergic reactions and anaphylaxis in animals and humans are attributed to MCs. In rodents, MCs are classified as mucosal and connective tissue types, whereas in humans, the MCs are classified on the basis of protease enzymes such as chymase (MCC), tryptase (MCT), or both chymase and tryptase (MCTC).2
It has been reported that a large number of MCs are present in human, rodent, and canine hearts. Human heart contains 90% of MCTC type mast cells.3 The physiological functions of MCs include angiogenesis, formation of Atrial natriuretic factor (ANF) from its precursor proANF and constitution of local renin-angiotensin system in the heart. The MCs are also involved in various cardiac pathological conditions such as ischemia-reperfusion injury, hypertension, cardiac arrhythmias, transplant rejections, myocarditis and hyperhomocysteinemia.4,5
The MCs are activated by a wide variety of stimuli such as interaction with immunoglobulin E (IgE) receptors, complement proteins, substance P, Reactive oxygen species (ROS), interleukin 33, endotoxins and lipopolysaccharides. When activated, the MCs rapidly release different bioactive chemical mediators stored in their granules into the local microenvironment. The degranulation of MCs does not cause their death and they are able to reconstitute their granules within few hours of degranulation by de novo synthesis.6 The main biologically active degranulation products are histamine, serotonin, adenosine, heparin, tryptase, chymase, cathepsin G, elastase, carboxy-peptidases and β-glucuronidase. The other products are platelet activating factor, prostaglandins, leukotrienes and thromboxane. In addition, MCs have also been reported to release Tumor necrosis factor-α (TNF-α) and growth factors like Transforming growth factor-β (TGF-β), nerve growth factor, vascular endothelial growth factor and fibroblast growth factor.2,7
ROLE OF MCs IN CARDIAC REMODELLING
Cardiac remodelling is defined as change in size, shape and function of the heart due to changes at molecular and cellular level in response to hemodynamic load, neurohormonal activation and some other factors.8 It composes various types of alterations including cardiac hypertrophy (concentric/eccentric), dilatation and myopathy. The early events such as cardiac hypertrophy are protective in nature to tackle stress on heart, however, the chronic exposure to stress leads to mal-adaptations such as dilatation and myopathy that consequently results in heart failure.9
While MCs are reported to perform some physiological functions as mentioned earlier, the proliferation of cardiac MCs and their recruitment from other organs has been reported in cardiac pathological conditions. The degranulation products of MCs such as histamine, TNF-α, TGF-β, renin and interleukins are purported to induce cardiac hypertrophy and heart failure. The degranulation of MCs and their increased density in tissues is considered to be inter-related. It has been postulated that various cytokines and chemokines released following MCs degranulation cause tissue injury, and consequently the affected area sends signals for the recruitment of more MCs from surrounding tissues, thereby leading to aggravated cardiac damage.
In spontaneously hypertensive rats, the MCs are reported to induce cardiac fibrosis and dysfunction and the treatment with a MC stabilizer, nedocromil, ameliorates fibrosis and ventricular dysfunction.10 Similarly, the use of MC stabilizing agents provide protection against myocarditis and diabetesinduced cardiomyopathy.11,12 The aorto-caval shunt induces volume overload on the heart and subsequently leads to cardiac remodelling. Volume overload-induced cardiac remodelling was more pronounced in normal rats than that of MC deficient rats. This observation emphasizes the deleterious role of MCs in the process of cardiac remodeling.13 Observation from our laboratory has indicated that increase in MC density is associated with hyperhomocysteinemia-induced cardiac hypertrophy and fibrosis in rats. Treatment with MC stabilizers, cromoglycate and ketotifen, markedly attenuated hyperhomocysteinemiainduced cardiac remodelling in rats.4
TNF-α is a cytokine produced by a variety of proinflammatory cells such as activated macrophages, natural killer cells, eosinophils, neutrophils and MCs. It has been observed that MCs are the major source for the production of TNF-α in the heart. Studies in experimental animals have suggested that TNF-α activates p38 Mitogen activated protein kinase (MAP kinase) and other hypertrophic genes, thereby causing cardiac dysfunction.14 The activation of MCs stimulates TNF-α/NFκB/ IL-6 cascade to induce cardiac hypertrophy.15 Interleukin-6 induces Janus kinase/Signal transducer and activator of transcription (JAK/STAT) pathway to cause cardiac hypertrophy in animal models.16 During myocardial ischemia-reperfusion injury, calcitonin gene related peptide and substance P lead to degranulation of MCs and activate local renin-angiotensin system.17 Chymase is a proteolytic enzyme present in MCs that facilitates the conversion of angiotensin-I to angiotensin-II and consequently evokes cardiac remodelling. A significant increase in chymase activity has been reported in various animal models of cardiac dysfunction. Based on these findings, chymase inhibitors are being designed to manage cardiac complications and are presently undergoing clinical trials for their cardio-protective activity. Some investigators have reported that estrogen hormone curtails formation of TNF-α and inhibits the release of chymase from MCs, thereby preventing TNF-α and chymase-mediated cardiac damage and fibrosis. This phenomenon seems to account for the lower occurrence of cardiac complications in premenopausal women in comparison to post-menopausal women and men.18,19 The MCs appear to activate local renin-angiotensin system in the heart that plays an important role in the induction of ventricular remodelling.17 Cathepsin D is another proteolytic enzyme that shares 60% homology with renin in its amino acid sequence, although it’s biological activity is far lesser than that of renin.17 Angiotensin-II and TNF-α enhance the production of Reactive oxygen species (ROS) by stimulating NADPH oxidase. The generated ROS further activate tyrosine kinase, protein kinase C and MAP kinase dependent hypertrophic pathways.4 Histamine released from MCs seems to enhance the expression of fetal genes such as c-fos, resulting in cardiac hypertrophy. The MCs-derived TGF-β causes proliferation of fibroblasts and promotes the synthesis of collagen in heart. It has been reported that TGF-β induces cardiac hypertrophy by up-regulating MAP kinase.20
The wide range of cardiac pathological factors which activate MCs and ensuing events leading to the release of biologically active products described in the foregoing discussion for cardiac remodelling are diagrammatically illustrated in Figure 1.
Figure 1: Diagrammatic Representation of the Activators and Released Products of Mast Cells Involved in Cardiac Remodelling
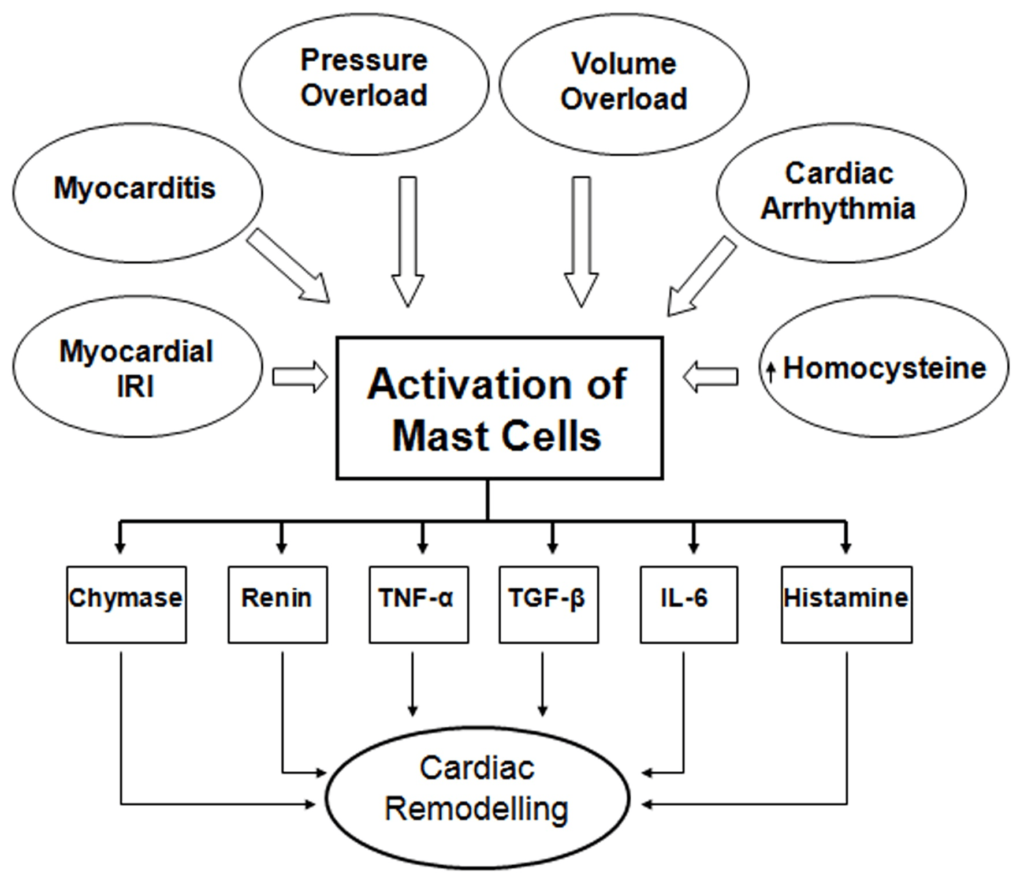
In conclusion, overwhelming evidence based on preclinical studies suggest that MCs are predominantly involved in causing cardiac remodelling and MC stabilizers such as cromoglycate, ketotifen and nedocromil ameliorate myocardial remodelling induced by a wide variety of pathologic stimuli in the heart. However, our understanding about how MCs are activated by factors shown in Figure 1 and consequently causing cardiac remodelling requires further investigations.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.


