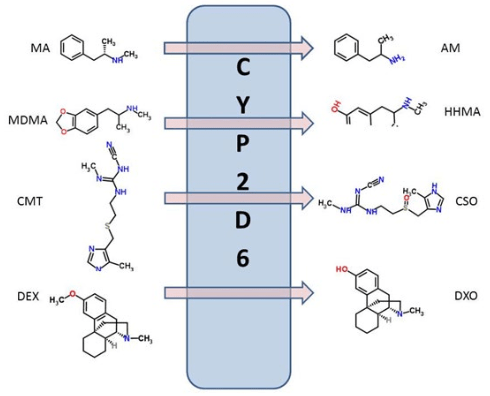
1. Badyal DK, Dadhich AP. Educational forum cytochrome P450 and drug interactions. Indian J Pharmacol. 2001; 33: 248-259.
2. Brown C. Overview of drug interactions modulated by cytochrome P450. US Pharm. 2001: HS26-HS45.
3. Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T. Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high sensitivity real time reverse transcription PCR. Yakugaku Zasshi. 2003; 123(5): 369-375. doi: 10.1248/yakushi.123.369
4. Drummer O, Odell M. Pharmacokinetics and duration of action. In: Bentliff G, McAllister L, eds. The Forensic Pharmacology of Drugs of Abuse. 1st ed. Abingdon, UK: Taylor & Francis; 2001: 27-29.
5. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013; 138(1): 103-141. doi: 10.1016/j.pharmthera.2012.12.007
6. Raunio H, Kuusisto M, Juvonen RO, Pentikäinen OT. Modeling of interactions between xenobiotics and cytochrome P450 (CYP) enzymes. Front Pharmacol. 2015; 6(123): 1-14. doi: 10.3389/fphar.2015.00123
7. Shen H, He MM, Liu H, et al. Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab Dispos. 2007; 35(8): 1292-1300. doi: 10.1124/dmd.107.015354
8. Qu Q, Qu J, Han L, et al. Inhibitory effects of phytochemicals on metabolic capabilities of CYP2D6*1 and CYP2D6*10 using cell-based models in vitro. Nat Publ Gr. 2014; 35(10): 685-696. doi: 10.1038/aps.2013.202
9. Cupp M, Tracy T. Cytochrome P450: New nomenclature and clinical implications. Am Fam Physician. 1998; 57(1): 107-116.
10. Van LM, Heydari A, Yang J, et al. The impact of experimental design on assessing mechanism-based inactivation of CYP2D6 by MDMA (Ecstasy). J Psychopharmacol. 2006; 20(6): 834-841. doi: 10.1177/0269881106062902
11. Van LM, Hargreaves JA, Lennard MS, Tucker GT, Rostami-Hodjegan A. Inactivation of CYP2D6 by methylenedioxymethamphetamine in different recombinant expression systems. Eur J Pharm Sci. 2007; 32(1): 8-16. doi: 10.1016/j.ejps.2007.05.002
12. Van LM, Swales J, Hammond C, Wilson C, Hargreaves JA, Rostami-Hodjegan A. Kinetics of the time-dependent inactivation of CYP2D6 in cryopreserved human hepatocytes by methylenedioxymethamphetamine (MDMA). Eur J Pharm Sci. 2007; 31(1): 53-61. doi: 10.1016/j.ejps.2007.02.005
13. Brown C. Overview of Drug–Drug Interactions with SSRIs. US Pharm. 2008: HS3-HS19.
14. Cupp MJ, Tracy TS. Cytochrome P450: New nomenclature and clinical implications. Am Fam Physician. 1998; 57(1): 107-116.
15. Martinez C, Albet C, Agundez JA, et al. Comparative in vitro and in vivo inhibition of cytochrome P450 CYP1A2, CYP2D6, and CYP3A by H2-receptor antagonists. Clin Pharmacol Ther. 1999; 65(4): 369-376. doi: 10.1016/S0009-9236(99)70129-3
16. Clemens KJ, Cornish JL, Li KM, Hunt GE, McGregor IS. MDMA (’Ecstasy’) and methamphetamine combined: Order of administration influences hyperthermic and long-term adverse effects in female rats. Neuropharmacology. 2005; 49(2005): 195-207. doi: 10.1016/j.neuropharm.2005.03.002
17. Maréchal J-D, Kemp C, Roberts G, Paine M, Wolf C, Sutcliffe M. Insights into drug metabolism by cytochromes P450 from modelling studies of CYP2D6-drug interactions. Br J Pharmacol. 2008; 153: 82-89. doi: 10.1038/sj.bjp.0707570
18. Drummer O, Odell M. Stimulants. In: Bentliff G, McAllister L, eds. The Forensic Pharmacology of Drugs of Abuse. 1st ed. Abingdon, UK: Taylor & Francis; 2001: 49-102.
19. Levine B. Principles of Forensic Toxicology. 4th ed. Washington, DC, USA: AACC Press; 2013.
20. Logan B. Methamphetamine-effects on human performance and behavior. Forensic Sci Rev. 2002; 14(1-2): 133-151.
21. Shoblock JR, Sullivan EB, Maisonneuve IM, Glick SD. Neurochemical and behavioral differences between d-methamphetamine and d-amphetamine in rats. Psychopharmacology (Berl). 2003; 165: 359-369. doi: 10.1007/s00213-002-1288-7
22. Logan B, Couper F. 3,4-Methylenedioxymethamphetamine — effects on human performance and behavior. Forensic Sci Rev. 2003; 15(1): 11-28.
23. Oesterheld JR, Armstrong SC, Cozza KL. Med-Psych Drug-Drug Interactions Update Ecstasy: Pharmacodynamic and Pharmacokinetic Interactions. Psychosomatics. 2004; 45(1): 84-87. doi: 10.1176/appi.psy.45.1.84
24. Casco C, Forcella MC, Beretta G, Grieco A, Campana G. Long-term effects of MDMA (Ecstasy) on the human central nervous system revealed by visual evoked potentials. Addict Biol. 2005; 10(2): 187-195. doi: 10.1080/13556210500123340
25. Escobedo I, O’Shea E, Orio L, et al. A comparative study on the acute and long-term effects of MDMA and 3,4-dihydroxymethamphetamine (HHMA) on brain monoamine levels after i.p. or striatal administration in mice. Br J Pharmacol. 2009; 144(2): 231-241. doi: 10.1038/sj.bjp.0706071
26. De Letter EA, Belpaire FM, Clauwaert KM, Lambert WE, Van Bocxlaer JF, Piette MH. Post-mortem redistribution of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”) in the rabbit. Part II: Post-mortem infusion in trachea or stomach. Int J Legal Med. 2002; 116(4): 225-232. doi: 10.1007/s00414-002-0293-z
27. Jantratid E, Prakongpan S, Dressman J, et al. Biowaiver monographs for immediate release solid oral dosage forms: Cimetidine. J Pharm Sci. 2006; 95(5): 974-984. doi: 10.1002/jps.20614
28. Liska DJ. The detoxification enzyme systems. Altern Med Rev. 1998; 3(3): 187-198.
29. Madeira M, Levine M, Chang TKH, Mirfazaelian A, Bellward GD. The effect of cimetidine on dextromethorphan O -demethylase activity of human liver microsomes and recombinant Cyp2D6. Drug Metab Dispos. 2004; 32(4): 460-467. doi: 10.1124/dmd.32.4.460
30. Park E, Cho H, Lee Y. Effect of Cimetidine and Phenobarbital on metabolite kinetics of Omeprazole in rats. Arch Pharm Res. 2005; 28(10): 1196-11202. doi: 10.1007/bf02972986
31. Draper AJ, Madan A, Parkinson A. Inhibition of coumarin 7-hydroxylase activity in human liver microsomes. Arch Biochem Biophys. 1997; 341(1): 47-61. doi: 10.1006/abbi.1997.9964
32. Suzuki T, Chiang H-JF, Misawa M. Effects of quinidine and cimetidine on the methamphetamine level in the rat brain. Jpn J Pharmacol. 1987; 43: 103-106. doi: 10.1254/jjp.43.103
33. Levine M, Bellward GD. Effect of cimetidine on hepatic cytochrome P450: Evidence for formation of a metabolite-intermediate complex. Drug Metab Dispos. 1995; 23(12): 1407-1411.
34. Larsson R, Erlanson P, Bodemar G, Norlander B, Fransson L, Strouth L. Pharmacokinetics of cimetidine and its sulphoxide metabolite during haemodialysis. Eur J Clin Pharmacol. 1982; 21(4): 325-330. doi: 10.1007/BF00637621
35. Ogbru O. Cimetidine, Tagamet HB. http://www.medicinenet.com/cimetidine/article.html. Accessed June 30, 2017.
36. Manap RA, Wright CE, Gregory A, et al. The antitussive effect of dextromethorphan in relation to CYP2D6 activity. Br J Clin Pharmacol. 1999; 48: 382-387. doi: 10.1046/j.1365-2125.1999.00029.x
37. CESAR. Dextromethorphan (DXM). Dextromethorphan (DXM). http://www.cesar.umd.edu/cesar/drugs/dxm.asp. Accessed June 30, 2017.
38. Nicholson K, Hayes B, Balster R. Evaluation of the reinforcing properties and phencyclidine-like discriminative stimulus effects of dextromethorphan and dextrorphan in rats and rhesus monkeys. Psychopharmacology (Berl). 1999; 146(1): 49-59. doi: 10.1007/s002130051087
39. Ehret GB, Daali Y, Chabert J, et al. Influence of CYP2D6 activity on pre-emptive analgesia by the N-Methyl-D-Aspartate antagonist dextromethorphan in a randomized controlled trial of acute pain. Pain Physician. 2013; 16: 45-56. doi: 10.36076/ppj.2013/16/45
40. Lotrich F, Rosen J, Pollock B. Dextromethorphan-induced delirium and possible methadone interaction. Am J Geriatr Pharmacother. 2005; 3(1): 17-20. doi: 10.1016/j.amjopharm.2005.03.002
41. Frank D, Jaehde U, Fuhr U. Evaluation of probe drugs and pharmacokinetic metrics for CYP2D6 phenotyping. Eur J Clin Pharmacol. 2007; 63(4): 321-333. doi: 10.1007/s00228-006-0250-8
42. Glick SD, Maisonneuve IM, Dickinson HA, Kitchen BA. Comparative effects of dextromethorphan and dextrorphan on morphine, methamphetamine, and nicotine self-administration in rats. Eur J Pharmacol. 2001; 422(1-3): 87-90. doi: 10.1016/S0014-2999(01)01066-4
43. Witherow LE, Houston JB. Sigmoidal kinetics of CYP3A substrates: An approach for scaling dextromethorphan metabolism in hepatic microsomes and isolated hepatocytes to predict in vivo clearance in rat. J Pharmacol Exp Ther. 1999; 290(1): 58-65.
44. Min DI, Ku Y-M, Vichiendilokkul A, Fleckenstein LL. A urine metabolic ratio of dextromethorphan and 3-methoxymorphinan as a probe for CYP3A activity and prediction of cyclosporine clearance in healthy volunteers. Pharmacotherapy1. 1999; 19(6): 753-759. doi: 10.1592/phco.19.9.753.31536
45. Kerryi NL, Somogyit AA, Bochner F, Mikus G, Somogyi AAA. The role of CYP2D6 in primary and secondary oxidative metabolism of dextromethorphan: In vitro studies using human liver microsomes. Br J clin Pharmac. 1994; 38(3): 243-248. doi: 10.1111/j.1365-2125.1994.tb04348.x
46. Magarey J. Dextromethorphan. INCHEM. http://www.inchem.org/documents/pims/pharm/pim179.htm. Published 1996. Accessed June 30, 2017.
47. Couper FJ, Logan BK. Drugs and Human Performance Fact Sheets – Methamphetamine and Amphetamines. 2014. https://www.nhtsa.gov/sites/nhtsa.dot.gov/files/809725-drugshumanperformfs.pdf. Accessed August 1, 2017.
48. Chauret N, Dobbs B, Lackman RL, et al. The use of 3- [ 2- ( N , N-Diethyl-N-Methylammonium ) Ethyl ] -7-Methoxy-4- methylcoumarin ( AMMC ) as a specific Cyp2D6 probe in Human liver microsomes. Drug Metab Dispos. 2001; 29(9): 1196-1200.
49. Riley RJ, Grime K. Metabolic screening in vitro: Metabolic stability, CYP inhibition and induction. Drug Discov Today Technol. 2004; 1(4): 365-372. doi: 10.1016/j.ddtec.2004.10.008
50. Crespi CL, Miller VP, Penman BW. Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes P450. Anal Biochem. 1997; 248(1957): 188-190. doi: 10.1006/abio.1997.2145
51. Palamanda JR, Favreau L, Lin C, Nomeir AA. Validation of a rapid microtiter plate assay to conduct cytochrome P450 2D6 enzyme inhibition studies. Drug Discov Today. 1998; 3(10): 466-470. doi: 10.1016/S1359-6446(98)01248-3
52. Turpeinen M, Korhonen LE, Tolonen A, et al. Cytochrome P450 (CYP) inhibition screening: Comparison of three tests. Eur J Pharm Sci. 2006; 29(2): 130-138. doi: 10.1016/j.ejps.2006.06.005
53. Nelson AC, Huang W, Moody DE. Variables in human liver microsome preparation: Impact on the kinetics of L-acetylmethadol (LAAM) N-demethylation and dextromethorphan O-demethylation. Drug Metab Dispos. 2001; 29(3): 319-325.
54. Florence VM, Di Stefano EW, Sum CY, Cho AK. The metabolism of (R)-(-)-amphetamine by rabbit liver microsomes. Drug Metab Dispos. 1982; 10(4): 312-315.
55. Bradford MM. A Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72: 248-254. doi: 10.1016/0003-2697(76)90527-3
56. Stresser DM, Turner SD, Blanchard AP, Miller VP, Crespi CL. Cytochrome P450 fluorometric substrates: Identification of isoform-selective probes for rat CYP2D2 and human CYP3A4. Drug Metab Dispos. 2002; 30(7): 845-852. doi: 10.1124/dmd.30.7.845
57. Pelkonen O, Puurunen J. Effect of cimetidine on microsomal drug metabolism in man. Biochem Pharmacol. 1980; 29(22): 3075-3080. doi: 10.1016/0006-2952(80)90448-7
58. Levine M, Law EY, Bandiera SM, Chang TK, Bellward GD. In vivo cimetidine inhibits hepatic CYP2C6 and CYP2C11 but not CYP1A1 in adult male rats. J Pharmacol Exp Ther. 1998; 284(2): 493-499.
59. Klotz U, Reimann IW. Drug Interactions through binding to cytochrome P450: The experience with H2-receptor blocking agents. Pharm Res. 1984; 1(2): 59-62.doi: 10.1023/A:1016347229923
60. Schmider J, Greenblatt DJ, Fogelman SM, Von Moltke LL, Shader RI. Metabolism of dextromethorphan in vitro: Involvement of cytochromes P450 2D6 and 3A3/4, with a possible role of 2E1. Biopharm Drug Dispos. 1997; 18(3): 227-240. doi: 10.1002/(SICI)1099-081X(199704)
61. Lin LY, Di Stefano EW, Schmitz DA, et al. Oxidation of methamphetamine and methylenedioxymethamphetamine by CYP2D6. Drug Metab Dispos. 1997; 25(9): 1059-1064.
62. Wu D, Victoria Otton S, Inaba T, Kalow W, Sellers EM. Interactions of amphetamine analogs with human liver CYP2D6. Biochem Pharmacol. 1997; 53(11): 1605-1612. doi: 10.1016/S0006-2952(97)00014-2
63. Sorkin E, Darvey D. Review of cimetidine drug interactions. Drug Intell Clin Pharmacol. 1983; 17(2): 110-120. doi: 10.1177/106002808301700205
64. Taavitsainen P, Kiukaanniemi K, Pelkonen O. In vitro inhibition screening of human hepatic P450 enzymes by five angiotensin-II receptor antagonists. Eur J Clin Pharmacol. 2000; 56(2): 135-140. doi: 10.1007/s002280050731
65. Yamamoto T, Suzuki A, Kohno Y. Application of microtiter plate assay to evaluate inhibitory EŠects of various compounds on nine cytochrome P450 isoforms and to estimate their inhibition patterns. Drug Metab Pharmacokinet. 2002; 17(5): 437-448. doi: 10.2133/dmpk.17.437
66. de la Torre R, Yubero-Lahoz S, Pardo-Lozano R, Farré M. MDMA, methamphetamine, and CYP2D6 pharmacogenetics: What is clinically relevant? Front Genet. 2012; 3(NOV): 1-8. doi: 10.3389/fgene.2012.00235
67. Orishiki M, Matsuo Y, Nishioka M, Ichikawa Y. In vivo administration of H2 blockers, cimetidine and ranitidine, reduced the contents of the cytochrome P450IID (CYP2D) subfamily and their activities in rat liver microsomes. Int J Biochem. 1994; 26(6): 751-758. doi: 10.1016/0020-711X(94)90104-X
68. Martinez C, Albet C, Agundez JA, et al. The genetic polymorphisms of drug-metabolizing enzymes are a common determinant of interindividual differences in the therapeutic effect and toxicity of many drugs. Clin Pharmacol Ther. 1999; 65(4): 369-376. doi: 10.1016/S0009-9236(99)70129-3
69. Rendić S. Drug interactions of H2-receptor antagonists involving cytochrome P450 (CYPs) enzymes: From the laboratory to the clinic. Croat Med J. 1999; 40(3): 357-367.
70. Ruffalo R, Thompson J, Segal J. Diazepam-cimetidine drug interaction: A clinically significant effect. South Med J. 1981; 74(9): 1075-1078. doi: 10.1097/00007611-198109000-00016
71. Dostalek M, Jurica J, Pistovcakova J, et al. Effect of methamphetamine on cytochrome P450 activity. Xenobiotica. 2007; 37(12): 1355-1366. doi: 10.1080/00498250701652877
72. Dostalek M, Hadasova E, Hanesova M, et al. Effect of methamphetamine on the pharmacokinetics of dextromethorphan and midazolam in rats. Eur J Drug Metab Pharmacokinet. 2005; 30(3): 195-201. doi: 10.1007/BF03190620
73. Yang PP, Huang EYK, Yeh GC, Tao PL. Co-administration of dextromethorphan with methamphetamine attenuates methamphetamine-induced rewarding and behavioral sensitization. J Biomed Sci. 2006; 13(5): 695-702. doi: 10.1007/s11373-006-9096-4
74. Kreth KP, Kovar KA, Schwab M, Zanger UM. Identification of the human cytochromes P450 involved in the oxidative Metabolism of “Ecstasy” – related designer drugs. Biochem Pharmacol. 2000; 59: 1563-1571. doi: 10.1016/S0006-2952(00)00284-7
75. Meyer MR, Peters FT, Maurer HH. The role of human Hepatic cytochrome P450 isozymes in the metabolism of racemic 3, 4-methylenedioxy- methamphetamine and its enantiomers. Pharmacology. 2008; 36(11): 2345-2354. doi: 10.1124/dmd.108.021543
76. Downey C, Daly F, O’Boyle KM. An in vitro approach to assessing a potential drug interaction between MDMA (ecstasy) and caffeine. Toxicol In Vitro. 2014; 28(2): 231-239. doi: 10.1016/j.tiv.2013.10.021
77. Shin EJ, Nah SY, Kim WK, et al. The dextromethorphan analog dimemorfan attenuates kainate-induced seizures via sigma1 receptor activation: comparison with the effects of dextromethorphan. Br J Pharmacol. 2005; 144(7): 908-918. doi: 10.1038/sj.bjp.0705998
78. Shin EJ, Nah SY, Chae JS, et al. Dextromethorphan attenuates trimethyltin-induced neurotoxicity via σ1 receptor activation in rats. Neurochem Int. 2007; 50(6): 791-799. doi: 10.1016/j.neuint.2007.01.008
79. Benetton SA, Fang C, Yang Y-O, et al. P450 Phenotyping of the metabolism of selegiline to desmethylselegiline and methamphetamine. Drug Metab Pharmacokinet. 2007; 22(2): 78-87. doi: 10.2133/dmpk.22.78
80. Cheng J, Zhen Y, Miksys S, et al. Potential role of CYP2D6 in the central nervous system. Xenobiotica. 2013; 43(11): 973-984. doi: 10.3109/00498254.2013.791410
81. Haduch A, Bromek E, Daniel WA. Role of brain cytochrome P450 (CYP2D) in the metabolism of monoaminergic neurotransmitters. Pharmacol Rep. 2013; 65(6): 1519-1528. doi: 10.1016/s1734-1140(13)71513-5
82. Ramamoorthy Y, Yu A, Suh N, Haining RL, Tyndale RF, Sellers EM. Reduced (±)-3,4-methylenedioxymethamphetamine (“Ecstasy”) metabolism with cytochrome P450 2D6 inhibitors and pharmacogenetic variants in vitro. Biochem Pharmacol. 2002; 63(12): 2111-2119. doi: 10.1016/S0006-2952(02)01028-6