1. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014; 64: 104-117. doi: 10.3322/caac.21220
2. World Health Organization. World Cancer Report 2014. NaturalezaCS; 2014.
3. Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007; 132: 2087-2102. doi: 10.1053/j.gastro.2007.03.052
4. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004; 4: 579-591. doi: 10.1038/nrc1408
5. Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010; 303: 235-241. doi: 10.1001/jama.2009.2014
6. Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999-2004. JAMA. 2006; 295: 1549-1555. doi: 10.1001/jama.295.13.1549
7. Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer. 2011; 11: 886-895. doi: 10.1038/nrc3174
8. Giovannucci E. Insulin and colon cancer. Cancer Causes Control. 1995; 6: 164-179. doi: 10.1007/bf00052777
9. Bardou M, Barkun AN, Martel M. Obesity and colorectal cancer. Gut. 2013; 62: 933-947. doi: 10.1136/gutjnl-2013-304701
10. Irrazabal T, Belcheva A, Girardin SE, Martin A, Philpott DJ. The multifaceted role of the intestinal microbiota in colon cancer. Mol Cell. 2014; 54: 309-320. doi: http://dx.doi.org/10.1016/j.molcel.2014.03.039
11. Aleman JO, Eusebi LH, Ricciardiello L, Patidar K, Sanyal AJ, Holt PR. Mechanisms of obesity-induced gastrointestinal neoplasia. Gastroenterology. 2014; 146: 357-373. doi: 10.1053/j.gastro.2013.11.051
12. Silventoinen K, Sans S, Tolonen H, et al. Trends in obesity and energy supply in the WHO MONICA Project. Int J Obes Relat Metab Disord. 2004; 28: 710-718. doi: 10.1038/sj.ijo.0802614
13. Wang Y, Monteiro C, Popkin BM. Trends of obesity and underweight in older children and adolescents in the United States, Brazil, China, and Russia. Am J Clin Nutr. 2002; 75: 971-977. doi: 10.1093/ajcn/75.6.971
14. Rennie KL, Jebb SA. Prevalence of obesity in Great Britain. Obes Rev. 2005; 6: 11-12. doi: 10.1111/j.1467-789X.2005.00164.x
15. Luo J, Hu FB. Time trends of obesity in pre-school children in China from 1989 to 1997. Int J Obes Relat Metab Disord. 2002; 26: 553-558. doi: 10.1038/sj.ijo.0801944
16. Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trends in obesity among US adults, 1999-2000. JAMA. 2002; 288: 1723-1727. doi: 10.1001/jama.2009.2014
17. Finkelstein EA, Khavjou OA, Thompson H, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med. 2012; 42: 563-570. doi: 10.1016/j.amepre.2011.10.026
18. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003; 348: 1625-1638. doi: 10.1056/NEJMoa021423
19. Le Marchand L, Wilkens LR, Kolonel LN, Hankin JH, Lyu LC. Associations of sedentary lifestyle, obesity, smoking, alcohol use, and diabetes with the risk of colorectal cancer. Cancer Res. 1997; 57: 4787-4794.
20. Murphy TK, Calle EE, Rodriguez C, Kahn HS, Thun MJ. Body mass index and colon cancer mortality in a large prospective study. Am J Epidemiol. 2000; 152: 847-854. doi: 10.1093/aje/152.9.847
21. Must A, Jacques PF, Dallal GE, Bajema CJ, Dietz WH. Long-term morbidity and mortality of overweight adolescents. A follow-up of the Harvard Growth Study of 1922 to 1935. N Engl J Med. 1992; 327: 1350-1355. doi: 10.1056/NEJM199211053271904
22. Thun MJ, Calle EE, Namboodiri MM, et al. Risk factors for fatal colon cancer in a large prospective study. J Natl Cancer Inst. 1992; 84: 1491-1500. doi: 10.1093/jnci/84.19.1491
23. IARC. Weight control and physical activity. IARC handbooks of cancer prevention. Lyon, France: IARC Press; 2002.
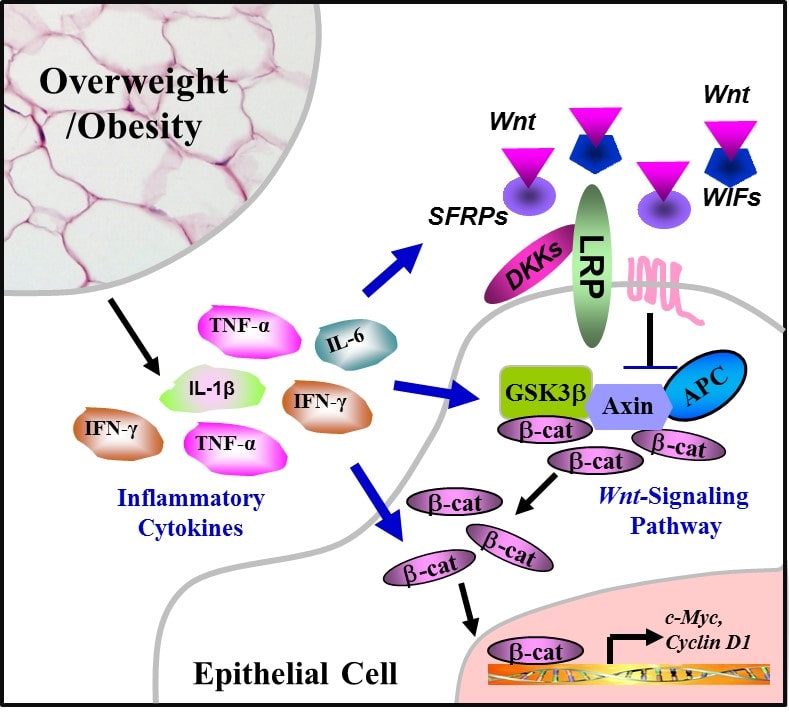
24. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993; 259: 87-91. doi: 10.1126/science.7678183
25. Fischer-Posovszky P, Wabitsch M, Hochberg Z. Endocrinology of adipose tissue – an update. Horm Metab Res. 2007; 39: 314-321. doi: 10.1055/s-2007-976539
26. Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006; 6: 772-783. doi: 10.1038/nri1937
27. Ramos EJ, Xu Y, Romanova I, et al. Is obesity an inflammatory disease? Surgery. 2003; 134: 329-335. doi: 10.1067/msy.2003.267
28. Ruan H, Zarnowski MJ, Cushman SW, Lodish HF. Standard isolation of primary adipose cells from mouse epididymal fat pads induces inflammatory mediators and down-regulates adipocyte genes. J Biol Chem. 2003; 278: 47585-47593. doi: 10.1074/jbc.M305257200
29. Howcroft TK, Campisi J, Louis GB, et al. The role of inflammation in age-related disease. Aging (Albany NY). 2013; 5: 84-93. https://doi.org/10.18632/aging.100531
30. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002; 420: 860-867. doi: 10.1038/nature01322
31. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001; 48: 526-535. doi: 10.1136/gut.48.4.526
32. Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 2007; 28: 1450-1459. doi: 10.1111/j.1745-7254.2007.00695.x
33. Zhang Y, Leung DY, Richers BN, et al. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol. 2012; 188: 2127-2135. doi: 10.4049/jimmunol.1102412
34. Spencer M, Finlin BS, Unal R, et al. Omega-3 fatty acids reduce adipose tissue macrophages in human subjects with insulin resistance. Diabetes. 2013; 62: 1709-1717. doi: 10.2337/db12-1042
35. Kim YS, Young MR, Bobe G, Colburn NH, Milner JA. Bioactive food components, inflammatory targets, and cancer prevention. Cancer Prev Res (Phila). 2009; 2: 200-208. doi: 10.1158/1940-6207.CAPR-08-0141
36. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2013; 140: 883-899. doi: 10.1016/j.cell.2010.01.025
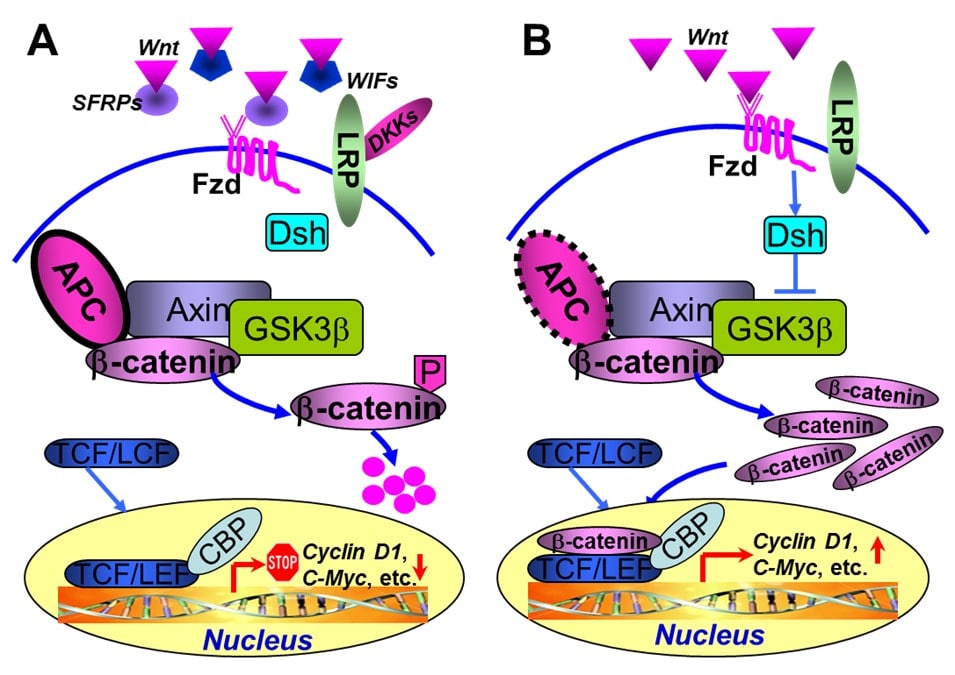
37. 2Taketo MM. Shutting down Wnt signal-activated cancer. Nat Genet. 2004; 36: 320-322. doi: 10.1038/ng0404-320
38. Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487: 330-337. doi: 10.1038/nature11252
39. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996; 87: 159-170. doi: 10.1016/S0092-8674(00)81333-1
40. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000; 103: 311-320. doi: 10.1016/S0092-8674(00)00122-7
41. White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/beta-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012; 142: 219-332. doi: 10.1053/j.gastro.2011.12.001
42. Suzuki H, Watkins DN, Jair KW, et al. Epigenetic inactivation of SFRP genes allows constitutive Wnt signaling in colorectal cancer. Nat Genet. 2004; 36: 417-422. doi: 10.1038/ng1330
43. Aguilera O, Fraga MF, Ballestar E, et al. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene. 2006; 25: 4116-4121. doi: 10.1038/sj.onc.1209439
44. Ying Y, Tao Q. Epigenetic disruption of the Wnt/beta-catenin signaling pathway in human cancers. Epigenetics. 2009; 4: 307-312.
45. Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci. 35: 161-168. doi: 10.1016/j.tibs.2009.10.002
46. Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006; 7: 885-896. doi: 10.1038/nrm2066
47. Christodoulides C, Lagathu C, Sethi JK, Vidal-Puig A. Adipogenesis and Wnt signalling. Trends Endocrinol Metab. 2009; 20: 16-24. doi: 10.1016/j.tem.2008.09.002
48. Ross SE, Hemati N, Longo KA, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000; 289: 950-953. doi: 10.1126/science.289.5481.950
49. Ouchi N, Higuchi A, Ohashi K, et al. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science. 2010; 329: 454-457. doi: 10.1126/science.1188280
50. Gustafson B, Smith U. Cytokines promote Wnt signaling and inflammation and impair the normal differentiation and lipid accumulation in 3T3-L1 preadipocytes. J Biol Chem. 2006; 281: 9507-9516. doi: 10.1074/jbc.M512077200
51. Cawthorn WP, Heyd F, Hegyi K, Sethi JK. Tumour necrosis factor-alpha inhibits adipogenesis via a beta-catenin/TCF4(TCF7L2)-dependent pathway. Cell Death Differ. 2007; 14: 1361-1373. doi: 10.1038/sj.cdd.4402127
52. Oguma K, Oshima H, Aoki M, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J. 2008; 27: 1671-1681. doi: 10.1038/emboj.2008.105
53. Liu Z, Brooks RS, Ciappio ED, et al. Diet-induced obesity elevates colonic TNF-alpha in mice and is accompanied by an activation of Wnt signaling: a mechanism for obesity-associated colorectal cancer. J Nutr Biochem. 2012; 23: 1207-1213. doi: 10.1016/j.jnutbio.2011.07.002
54. Kaler P, Augenlicht L, Klampfer L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: a crosstalk interrupted by vitamin D3. Oncogene. 2009; 28: 3892-3902. doi: 10.1038/onc.2009.247
55. Nava P, Koch S, Laukoetter MG, et al. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity. 2010; 32: 392-402. doi: 10.1016/j.immuni.2010.03.001
56. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011; 121: 2111-2117. doi: 10.1172/JCI57132