1. Kochar Kaur K, Allahabadia GN, Singh M. An update on Aetiopathogenesis and management of obesity. Obes Control Ther. 2016; 3(1): 1-17. doi: 10.15226/2374-8354/2/2/00123
2. Kochar Kaur K, Allahabadia GN, Singh M. Current management of obesity in an infertile female. Recent Advances and Future Prospectives. Drugs J Pharm Nutr Soc. 2013; 3: 1-13.
3. Kochar Kaur K, Allahabadia GN, Singh M. Further update on the management of obesity with emphasis on genetic perspective. BAOJ Obe Weigh Manage. 2016; 3(1): 1-17.
4. Kochar Kaur K, Allahabadia GN, Singh M. A review on Nutrient Metabolism with special emphasis on fatty acid metabolism. BAOJ Food Sci Tec. 2017; 1: 1: 001.
5. Kochar Kaur K, Allahabadia GN, Singh M. Therapeutic implications of the recent understanding of brown or beige adipocyte physiology. Adv Tech Biol Med. 2016; 3: 128. doi: 10.4172/2379-1764.1000128
6. Kochar Kaur K, Allahabadia GN, Singh M. Recent advances in BAT/beige physiology-current therapeutic applications. J Diab and Metab Control. 2016.
7. Kochar Kaur K, Allahabadia GN, Singh M. An update on miR’s in obesity and metabolic dieases-A review. Metabolomics. 2014.
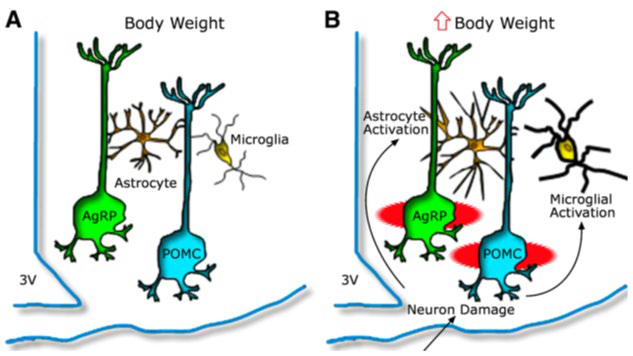
8. Kochar Kaur K, Allahabadia GN, Singh M. Hypothalamic inflammation and gliosis in obesity a etiopathogenesis specially with high fat diet. Proceedings of the World Congress on and Obesity Diabetes and cardiology; 10-12th July, 2017; Dubai, UAE.
9. Thaler JP, Yi CX, Schur EA, et al. Obesity is accociated with hypothalamic injury in rodents and humans. J Clin Invest. 2012; 122(1): 153-162. doi: 10.1172/JCI59660
10. Lee D, Thaler JP, Berseth KE, Melhorn SJ, Schwartz MW, Schur EA. Longer T2 relaxation timeis a marker of hypothalamic gliosis in mice with diet induced obesity. Am J Physiol Endocrinol Metab. 2013; 304(11): E1245-E1250. doi: 10.1152/ajpendo.00020.2013
11. Gyenet SJ, Nguyen HT, Hwang BH, Schwartz MW, Baskin DG, Thaler JP. High fat diet feeding causes rapid, non apoptotic cleavage of caspase 3 in astrocytes. Brain Res. 2013; 1512: 97-105. doi: 10.1016/j.brainres.2013.03.033
12. Garcia-Caceres C,Yi CX, Tschop MH. Hypothalamic astrocytes in obesity. Endocrinol Metab Clin North Am. 2013; 42(1): 57-66. doi: 10.1016/j.ecl.2012.11.003
13. Morari J, Anhe GF, Nascimento LF, et al. Fractalkaline (CX3CL1) is involved in the early activation of experimental obesity. Diabetes. 2014; 63(11): 3770-3784. doi: 10.2337/db13-1495
14. Valdearcos M, Xu AW, Koliwad SK. Hypothalamic inflammation in the control of metabolic function. Annu Rev Physiol 2015; 77: 131-160. doi: 10.1146/annurev-physiol-021014-071656
15. Myers MG Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and insulin resistance: Disringuishing cause from effect. Trends Endocrinol Metab. 2010; 2(11): 643-651. doi: 10.1016/j.tem.2010.08.002
16. Parton LE, Ye CP, Coppari R, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007; 449(7159): 228-232. doi: 10.1038/nature06098
17. Kumar RB, Arrone LJ. Hypothalamic inflammation: Is there evidence for human obesity? Curr Obes Rep. 2014; 3(2): 242-247. doi: 10.1007/s13679-014-0104-0
18. Briellmann RS, Kalnis RM, Berkovic SF, Jackson GD. Hippocampal pathology in refractory temporal lobe epilepsy: T2 weighted signal changes reflect dentate gliosis. Neurology. 2002; 58(2): 265-271.
19. Braffman BH, Zimmerman RA, Trojanowski JQ, Gonates NK, Hickey WF, Schlapfer WW. Brain MR: Pathologic coorelations with gross and histopathology. 2. Hyperintense white matter foci in the elderly. AJR Am J Roentgenol. 1988; 151(3): 559-566. doi: 10.2214/ajr.151.3.559
20. Marshall VG, Bradley WG Jr, Marshal CE, Bhoopat T, Rhodes RH. Deep white matter infarction: Correlation of MR imaging and histopathologic findings. Radiology. 1988; 167(2): 517-522. doi: 10.1148/radiology.167.2.3357964
21. Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the Mcdonald Criteria.Ann Neurol. 2005; 58(6): 840-846. doi: 10.1002/ana.20703
22. Third report of the National Cholesterol Education Program (NCEP) Expert panel on Detection, Evaluation and treatment of high blood cholesterol in adults (Adult treatment Panel III) final report. Circulation. 2002; 106(25): 3143-3421.
23. Schur EA, Melhorn SJ, Oh SK, et al. Radiologic evidence that hypothalamic gliosis is associated with obesity and insulin resistance in humans. Obesity (Silver Spring). 2015; 23(11): 2142-2148. doi: 10.1002/oby.21248
24. Douglass JD, Dorfman MD, Thaler JP. Glia: Silent partners in energy homeosrasis and obesity pathogenesis. Diabetolgia. 2017; 60(2): 226-236. doi: 10.1007/s00125-016-4181-3
25. Buckman LB, Thomson MM, Moreno HN, Ellacott KLJ. Regional astrogliosis in the mouse hypothalamus in response to obesity. J Comp Neurol. 2013; 521(6): 1322-1333. doi: 10.1002/cne.23233
26. Buckman LB, Hasty AH, Flaherty DK, et al. Obesity induced by a high fat dirt is associated with increased immune cellentry into the central nervous system. Brain Behav Immun. 2014; 35: 33-42. doi: 10.1016/j.bbi.2013.06.007
27. Berkseth KE, Guyenet SJ, Melhorn SJ, et al. Hypothalamic glioses associated with high fat diet feeding is reversible in mice: A combined immunohistochmical and magnetic resonance imaging study. Endocrinology. 2014; 155(8): 2858-2867. doi: 10.1210/en.2014-1121
28. Andre C, Guzman-Quevedo O, Rey C, et al. Inhibiting the microglia expansion prevents diet induced hypothalamic and peripheral inflammation. Diabetes. 2017; 66(4): 908-919. doi: 10.2337/db16-0586
29. Buckman LB,Thomson MM, Lippert RN, Blackwell TS, Yuli FE, Ellacott KLJ. Evidence for a novel functional role of astrocytes in the acute homeostatic response to high fat diet intake in mice. Molecular Metab. 2015; 4(1): 58-63. doi: 10.1016/j.molmet.2014.10.001
30. Sofroniew MV, Vinters HV. Astrocytes: Biologyand pathology. Acta Neuropathologia. 2010; 119(1): 7-35. doi: 10.1007/s00401-009-0619-8
31. Yamanaka K, Chun SJ, Bollee S, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008; 11(3): 251-253. doi: 10.1038/nn2047
32. Osborn LM, Kamphuis W, Wadman WJ, Hol EM. Asitrogliosis: An integral player in the pathogenesis of Alzheimers disease. Prog Neurobiol. 2016; 144: 121-141. doi: 10.1016/j.pneurobio.2016.01.001
33. Yang L, Qi Y, Yang Y. Astrocyes controlfood intake by inhibiting AgRP neuron activity via adehisine A1 receptors. Cell Reports. 2015; 11(5): 798-807. doi: 10.1016/j.celrep.2015.04.002
34. Chen N, Sugihara H, KimJJJ, et al. Direct modulation of GFAP expressing glia in the arxuate nucleus biderectionally regulates feeding. eLife. 2016; 5: 5599-5609. doi: 10.7554/eLife.18716
35. Kim JG, Suyama S, Koch M, et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuitsand feeding. Nature Neurosci. 2014; 17(7): 908-910. doi: 10.1038/nn.3725
36. Fuente-Martin E, Garcia-Ciceres C, Argentine-Arizon P, et al. Ghrelin regulates glucose and glutamate transporters in hypothalamic astrocytes. Science Reports. 2016; 6: 23673. doi: 10.1038/srep23673
37. Reiner DJ, Mietlicki Baase EG, Mcgrath LE, et al. Astrocytes regulate GLP1 receptor mediated effects on energy balance. J Neuroscience. 2016; 36(12): 3531-3540. doi: 10.1523/JNEUROSCI.3579-15.2016
38. Garcia-Ciceres C, Quarta C, Varela L, et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell. 2016; 166(4): 867-880. doi: 10.1016/j.cell.2016.07.028
39. Douglass JD, Dorfman MD, Fasnacht R, Schaffer LD, Thaler JP. Astrocyte IKKβ/NFĸB signaling is required for diet induced obesity and hypothalamic inflammation. Molecular Metab. 2017; 6(4): 366-373. doi: 10.1016/j.molmet.2017.01.010
40. Yang Y, Zhou IG, Wu QJ, Ma X, Xiang YB. Association between dietary fiber and lower risk of all cause mortality: A metaanalysis of cohort studies. Am J Epidemiol. 2015; 181(2): 83-91. doi: 10.1093/aje/kwu257
41. Slavin R. Dietary fiberand body weight. Nutrition. 2005; 21(3): 411-418. doi: 10.1016/j.nut.2004.08.018
42. Wanders AJ, Van den Borne JJ, et al. Effects of dietary fibre on subjective appetite, energy intake and body weight: A systematic review of randomized controlled trials. Obes Rev. 2011; 12(9): 724-739. doi: 10.1111/j.1467-789X.2011.00895.x
43. Lattimer JM, Haulo MD. Effect of dietary fiber and its components on metabolic health. Nutrients. 2010; 2(12): 1266-1289. doi: 10.3390/nu2121266
44. Guynet SJ, Schwatz MJ. Regulation of food intake energy balance and body fat mass: Implications for the pathogenesis and treatment of obesity. J Clin Endocrinol Metab. 2012; 97(3): 745-755. doi: 10.1210/jc.2011-2525
45. Malijaars P, Peters H, Mela D, Masclee A. A ilieal break: A sensible food target for appetite control.A review. Physiol Behav. 2008; 95(3): 271-281. doi: 10.1016/j.physbeh.2008.07.018
46. Murphy KG, Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature. 2006; 444(7121): 854-859. doi: 10.1038/nature05484
47. Nilsson AC, Johanson–Boll EV, Bjorck IME. Increased gut hormones and insulin sensitivityindex following a 3-d ntervention with barleykernel based product: A randomized cross over study inhealth middle aged subjects. Br J Nutr. 2015; 114(6): 899-907. doi: 10.1017/S0007114515002524
48. Ye Z, Arumugam V, Haugabrooks E, Williamson P, Hendrich S. Soluble dietary fibre (Fibersol-2) decreases hungerand increases satiety hormonesin humans when ingested with a meal. Nutr Res. 2015; 35(5): 393-400. doi: 10.1016/j.nutres.2015.03.004
49. Sarda FAH, Giuntini FB, Gomez ML, et al. Impact of resistant starch from unripe banana flour on hunger, satiety and glucose homeostasis in healthy volunteers. J Func Foods. 2016; 24: 63-74. doi: 10.1016/j.jff.2016.04.001
50. Vasselli JR, Scarpace PJ, Harris RBS, Banks WA. Dietary components in the development of insulin resistance. Adv Nutr. 2013; 4: 164-175. doi: 10.3945/an.112.003152
51. Robertson MD, Bickerton AS, Dennis AI, Vidal H, Frayn KN. Insulin sensitizing effect of dietary resistant starch and effects on skeletal muscleand adipose tissue metabolism. Am J Clin Nutr. 2005; 82(3): 559-567.
52. Maki KC, Pelkman CL, Finocchiaro ET, et al. Resistant starch from high amylase maize increases insulin sensitivity in overweight and obese men. J Nutr. 2012; 42(4): 717-723. doi: 10.3945/jn.111.152975
53. Robertson MD, Wright JW, Loizon E, et al. Insulin sensitizing effects on muscle and adipose tissue after dietary fiber intake in men and women with metabolis syndrome. J Clin Endocrinol Metab. 2012; 297(9): 3326-3332. doi: 10.1210/jc.2012-1513
54. Johnston K, Thomas E, Bell J, Frost G, Robertson M. Resistant starch improves insulin sensitivity in metabolic syndrome.Diab Med. 2010; 27(4): 391-397. doi: 10.1111/j.1464-5491.2010.02923.x
55. Higgins JA, Higbee DR, Donahoo WT, Brown IL, Bell ML, Bessett DH. Resistant starch consumption promotes lipid oxidation. Nutr Met. 2004; 1(1): 8. doi: 10.1186/1743-7075-1-8
56. Ebhihara K, Shiraishi R, Okuma K. Hydroxy propyl-modified potato starch increases fecalbile acid excretion in rats. J Nutr. 1998; 128: 848-854.
57. Venkatraman A, Sieber JR, Schmidt AW, et al. Variable responses of human microbiomes to dietary supplementation with resistant starch. Microbiome. 2016; 4: 33. doi: 10.1186/s40168-016-0178-x
58. Maziarz MP, Preisendanz S, Juma S, Imrhan V, Prasad C, Vijayagopal P. Resitant starch lowers postprandial glucose and leptin in overweight adults consuming a moderate to high fat diet: A randomized controlled trial. Nutr Journal. 2017; 16: 14. doi: 10.1186/s12937-017-0235-8
59. Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPAR alpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007; 5(6): 426-437. doi: 10.1016/j.cmet.2007.05.002
60. Inagaki T, Dutchak P, Zhao G, et al. Endocrine regulation of the fasting response by PPAR alpha mediated induction of fibroblast growth factor 21. Cell Metab. 2007; 5: 415-425. doi: 10.1016/j.cmet.2007.05.003
61. Potthoff MJ, Inagaki T, Satapati S, et al. FGF21 induces PGC-1 alpha and regulates carbohydratesand fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci USA. 2009; 106: 10853-10858. doi: 10.1073/pnas.0904187106
62. Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factor 15/19 and 21 from feast to famine. Genes Dev. 2012; 26: 312-324. doi: 10.1101/gad.184788.111
63. Laeger T, Henagan TM, Albarado DC, et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest. 2014; 124: 3913-3922. doi: 10.1172/JCI74915
64. DeSouza–Coelho AL, Marrero PF, Haro D. Activating transcription factor4 dependent induction of FGF21 during amino acid deprivation. Biochem J. 2012; 443: 165-171. doi: 10.1042/BJ20111748
65. Wek SA, Zhu S, Wek RC. The histidyl-tRNA synthetase related sequencein the Eif2alpha protein kinase GCN2 interacts with Trna and is required for activation in response to starvation for different amino acids. Mol Cell Biol. 1995; 15: 4497-4506. doi: 10.1128/MCB.15.8.4497
66. Xiao F, Huang Z, Li H, et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/Mtor/S6K1 and AMPK pathways. Diabetes. 2011; 60: 746-756. doi: 10.2337/db10-1246
67. Zhang P, McGrath BC, Reinert J, et al. The GCN2Eif2 alpha kinase is required for adaptation to amino acid deprivationin mice. Mol Cell Biol. 2002; 22: 6681-6688. doi: 10.1128/MCB.22.19.6681-6688.2002
68. Kim KH, Jeong YT, Kim SH. Metformin induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation. Biochem Biophys Res Commun. 2013; 440: 76-81. doi: 10.1016/j.bbrc.2013.09.026
69. Schaap HG, Kremer AE, Lamers WH, Jansen PL, Gaemers IC. Fibroblast growth factor 21 is induced by endoplasmic reticulum stress. Biochimie. 2013; 95: 692-699. doi: 10.1016/j.biochi.2012.10.019
70. Wilson GJ, Lennox BA, She P, et al. GCN2 is required to increase fibroblast growth factor 21 and maintain hepatic triglyceride homeostasis during asparaginase treatment. Am J Physiol Endocrinol Metab. 2015; 308(4): E283-E293. doi: 10.1152/ajpendo.00361.2014
71. Laegar T, Albarado DC, Burke SJ, et al. Metabolic responses to dietary protein restriction require an increasein FGF21 that is delayed in absence of GCN2. Cell Rep. 2016; 16(3): 707-716.doi: 10.1016/j.celrep.2016.06.044
72. Hartmann P, Ramseier A, Gundat F, Mihatsch MJ, Polasek W. Normal weight of the brain in adults in relation to age, sex body height and weight. Pathologie. 1994; 15: 165-170.
73. Peters A, Schweiger U, Pellerin L, et al. The selfish brain: Competition for energy resources. Neurosci Behav Rev. 2004; 28: 143-180. doi: 10.1016/j.neubiorev.2004.03.002
74. Sokoloff L, Reivich M, Kennedy C, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anaesthetized albino rat. J Neurochem. 1977; 23: 897-916. doi: 10.1111/j.1471-4159.1977.tb10649.x
75. Edmond J. Energy metabolism in developing brain cells. Can J Physiol Pharmacol. 1992; 70(S1): S118-S129. doi: 10.1139/y92-253
76. Yi CX, Habegger KM, Chowen JA, Stern J, Tscop MH. Arole for astrocytes in the central control of metabolism. Neuroendocrinology. 2011; 93: 143-149. doi: 10.1159/000324888
77. Abbott NJ, Revest PA, Romero IA. Astrocyte oendothelial interaction: Physiology and pathology. Neuropathol Appl Neurobiol. 1992; 18: 424-433. doi: 10.1111/j.1365-2990.1992.tb00808.x
78. Tsacopolous M, Magistretti PJ. Metabolic coupling between glia and neurons. J Neurosci. 1996; 16: 877-885.
79. Liu B, Niu L, Shen MZ, et al. Decreased astroglial monocarboxylate transporter 4 expression in temporal lobe epilepsy. Mol Neurobiol. 2014; 50: 327-338.
80. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A; 91: 10625-10629.
81. Perea G, Navarette M, Araque A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci; 32(8): 421-431. doi: 10.1016/j.tins.2009.05.001
82. Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. J Cereb Blood Flow Metab. 2012; 32: 1152-1166. doi: 10.1038/jcbfm.2011.149
83. Pellerin L, Pellegri G, Bittar PG, et al. Evidence supporting the existence of an activity dependent astrocyte–neuron lactate shuttle. Dev Neurosci. 1998; 20: 291-299.
84. Blanger M, Allaman I, Magistretti PJ. Brain energy metabolism: Focus on astrocyte–neuron metabolic cooperation. Cell Metab. 2011; 14: 724-738. doi: 10.1016/j.cmet.2011.08.016
85. LeFoll C, Dunn Meynell AA, Miziorko HM, Levin BE. Regulation of hypothalamic neuronal sensing and food intake by ketone bodies and fatty acids. Diabetes. 2014; 63: 1259-1269. doi: 10.2337/db13-1090
86. LeFoll C, Dunn Meynell AA, Miziorko HM, Levin BE. Role of VMH ketone bodiesin adjusting caloric intaketo increased dietary fat content in DIO and DR rats. Am J Physiol Regul Integr Comp Physiol. 2015; 308: R872-R878. doi: 10.1152/ajpregu.00015.2015
87. Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: Focus on astrocyte neuron metabolic cooperation. Cell Metab. 2011; 14: 724-738. doi: 10.1016/j.cmet.2011.08.016
88. LeFoll C, Levin BE. Fatty acid-induced astrocyte ketone production and the control of food intake. Am J Physiol Regul Integr Comp Physiol. 2016; 310(11): R1186-R1192. doi: 10.1152/ajpregu.00113.2016
89. Serhari CN, Savill J. Resilution of inflammation: The beginning programs the end. Nat Immunol. 2005; 6: 191-197. doi: 10.1038/ni1276
90. Takano T, Clish CB, Gronert K, Pertasis N, Serhan CN. Neutrophil mediated changes in vascular permeabilityare inhibited by topical applications of asprin triggered 15-epi-lipoxin A4 and novel lipoxin B4 stable analogues. J Clin Invest. 1998; 101: 819-826. doi: 10.1172/JCI1578
91. Serhan CN, Hong S, Gronert K, et al. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by asprin treatment that counter proinflammatory signals. J Exp Med. 2002; 106: 1025-1037.
92. Arita M, Bianchini J, Alberti J, et al. Stereochemical assignment ,anti-inflammatory properties and receptor for omega3 lipid mediator resolvin E1. J Exp Med. 2005; 201: 713-722. doi: 10.1084/jem.20042031
93. Serhan CN, Gotlinger K, Hong S, et al. Resolving inflammation: Dual antiinflammatory actions of neuroprotectin D1/Protectin D1 and its natural stereoisomer assignments of dihydroxy containing docosatrienes. J Immunol. 2006; 176: 1848-1859.
94. Serhan CN, Chang N, Van Dyke TE. Resolving inflammation: Dual antiinflammatory and proresolution lipid mediators.Nat Rev Immunol. 2008; 8: 349-361. doi: 10.1038/nri2294
95. Chung H, Fredman G, Backhed F, et al. Infection regulates proresolving mediators that lower antibiotic requirements. Nature. 2012; 484: 524-528.
96. Norling IV, Dalli J, Flower RJ, Serhan CN, Perretti M. Resolvin D1 limits polymorphonuclear leukocytes-recruitment to inflammatory loci: Receptor dependent actions. Arterioscler Thromb Vasc Biol. 2012; 232: 1970-1978. doi: 10.1161/ATVBAHA.112.249508
97. Claria J, Dalli J, Yacoubian S, Gao F, Serhan CN. Resolvin D1 and resolvin D2 govern local inflammatory tone in obese fat. J Immunol. 2012; 189: 2597-2605. doi: 10.4049/jimmunol.1201272
98. Neunofer A, Zeyda M, Mascher D, et al. Impaired local production of proresolving lipid mediators in obesity and 17HDHA as a potential treatment for obesity associated inflammation. Diabetes. 2013; 62: 1945-1956. doi: 10.2337/db12-0828
99. Moraes JC, Coope A, Morari J, et al. High fat diet induces apoptosis of hypothalamic neurons. PLoS One. 2009; 4: e5045. doi: 10.1371/journal.pone.0005045
100. Milanski M, Dejasperi G, Coope A, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J Neurosci. 2009; 29: 359-370.doi: 10.1523/JNEUROSCI.2760-08.2009
101. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKK beta /NF kappa B and ER stress links overnutrition to energy balancend obesity. Cell. 2008; 13561-13573. doi: 10.1016/j.cell.2008.07.043
102. DeSouza-CT, Araujo EP, Bordin S, et al. Consumption of a fat rich diet activates a proinflammatory response and induces insulin resistancein the hypothalamus. Endocrinology. 2005; 146: 4192-4199. doi: 10.1210/en.2004-1520
103. Velloso LA, Folli F, Saad MJ. TLR4 at the crossroads of nutrients, gut microbiotaand metabolic inflammation. Endocr Rev. 2015; 36: 245-271. doi: 10.1210/er.2014-1100
104. Ignacio-Souza LM, Bonbassaro B, Pascol LB, et al. Defective regulation of the ubiquitin/proteasome system in the hypothalamus of obese male mice. Endocrinology. 2014; 155: 2831-2844. doi: 10.1210/en.2014-1090
105. Romanatto T, Roman EA, Arruda AP, et al. Deletion of tumor necrosis factor alpha receptor (TNFR1) protects against diet induced obesity by means of increased thermogenesis. J Biol Chem. 2009; 284: 36213-36222. doi: 10.1074/jbc.M109.030874
106. Cintra DE, Ropelle ER, Moraes JC, et al. Unsaturated fatty acids reverse diet induced hypothalamic inflammation in obesity. PLoS One. 2012; 7: e30571. doi: 10.1371/journal.pone.0030571
107. Nascimento LF, Souza GF, Morari J, et al. Omega 3 fatty acids induce neurogenesis of predominantly POMC expressing cells in the hypothalamus. Diabetes. 2016; 65: 673-686. doi: 10.2337/db15-0008
108. Pascoal LB, Bombasarro B, Ramalho AF, et al. Resolvin RvD2 reduces hypothalamic inflammation and rescues mice from diet induced obesity. J Neuroinflammation. 2015; 14: 5. doi: 10.1186/s12974-016-0777-2
109. Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J obese (Lond). 2008; 32(Suppl 7): 552-554. doi: 10.1038/ijo.2008.238
110. Hill JQ, Peters JC. Environmental contributions to the obesity epidemic. Science. 1998; 280: 1371-1374. doi: 10.1126/science.280.5368.1371
111. Flier JS. Obesity wars: Molecular progress confronts an expanding epidemic. Cell. 2004; 116: 337-350. doi: 10.1016/S0092-8674(03)01081-X
112. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunityand fatty acid induced insulin resistance. J Clin Invest. 2006; 116: 3015-3025. doi: 10.1172/JCI28898
113. Velloso LA, Fiznick DL, Croop M. Type2 diabetes mellitus-An autoimmune disease? Nat Rev Endocrinol. 2013; 9: 750-755. doi: 10.1038/nrendo.2013.131
114. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012; 18: 363-374. doi: 10.1038/nm.2627
115. Velloso LA, Shwartz MW. Altered hypothalamic function in diet induced obesity. Int J obese(Lond). 2011; 35: 1455-1465. doi: 10.1038/ijo.2011.56
116. Fiznick DE, Cardozo AK, Crop M. The role for endoplasmic reticulum stressin diabetes mellitus. Endocr Rev. 2008; 29: 42-61.
117. Crop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesityand diabetes. Trends Mol Med. 2012; 18: 59-68. doi: 10.1016/j.molmed.2011.07.010
118. Samuel VT, Schulmn GI. Mechanisms for insulin resistance: Common threads and missing links. Cell. 2012; 148: 852-871. doi: 10.1016/j.cell.2012.02.017
119. Donathan MY, Shoelson SE. Type 2 diabetes mellitus as an inflammatory disease. Nat Rev Immunol. 2011; 11: 98-107. doi: 10.1016/j.diabres.2006.06.007
120. Cregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011; 29: 415-445. doi: 10.1146/annurev-immunol-031210-101322
121. McNeils JC, Olefsky JM. Macrophages, immunity and metabolic disease. Immunity. 2014; 41: 36-48. doi: 10.1016/j.immuni.2014.05.010
122. Goldfine AB, Fonseca V, Jablonski KA, et al. Targeting inflammation using salsalate in patients with type 2 diabetes: A randomized trial. Annu Intern Med. 2013; 159: 1-12. doi: 10.2337/dc13-0859
123. Nagasumi K, Esaki R, Iwachidow K, et al. Overexpression of GPR40 in pancreatic beta cells, augments glucose –stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes. 2009; 58: 1067-1076. doi: 10.2337/db08-1233
124. Oh DY, Takukdar S, Kae EJ, et al. GPR120 ia an omega 3 fatty acid receptor mediating potent anti-inflammatory and insulin sensitizing effects. Cell. 2010; 142: 687-698. doi: 10.1016/j.cell.2010.07.041
125. Cinitra DE, Ropelle ER, Moraes JC, et al. Unsaturated fatty acidsrevert diet induced hypothalamic inflammation in obesity.PLoS One. 2012; 7: e30571. doi: 10.1371/journal.pone.0030571
126. Itoh Y, Kuwamata Y, Harada M, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40.Nature. 2003; 422: 173-176. doi: 10.1038/nature01478
127. Oh DY, Olefsky JM. Omega 3 fatty acids and GPR120. Cell Metab. 2012; 15: 564-565. doi: 10.1016/j.cmet.2012.04.009
128. Da Oh Y, Walenta I, Akiyama TE, et al. GPR120 selective agonists improves insulin resistance and chronic inflammation in obese mice. Nat Med. 2014; 20: 942-947. doi: 10.1038/nm.3614
129. Burant CF. Activation of GPR40 as a therapeutic target for the treatment of diabetes. Diabetes Care. 2013; 36 (Suppl 2): 5175-5179. doi: 10.2337/dcS13-2037
130. Briscoe CP, Tadayyon M, Andrews JL, et al. The orphan G protein coupled receptor GPR40 is activated by median and long chain fatty acids. J Biol Chem. 2003; 278: 11303-11311.
131. Briscoe CP, Peat AJ, Mckeown SC, et al. Pharmacological regulation of insulin secretion in MIN 6 cells through the fatty acid eceptor GPR40: Identification of agonist and antagonist small molecules. Br J Pharmacol. 2006; 148: 619-628. doi: 10.1038/sj.bjp.0706770
132. Fdfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acidsstimulation of incretin secretion. Diabetes. 2008; 57(9): 2280-2287. doi: 10.2337/db08-0307
133. Boneva NB, Yamashima T. New insights into GPR40-CREB interactions in adult neurogenesis specific for primates. Hippocampus. 2012; 22(4): 296-305. doi: 10.1002/hipo.20951
134. Dragano NRV, Solon C, Ramalho AF, et al. Polyunsaturated fatty acids receptors, GPR40 and GPR120, are expressed in the hypothalamus and control energy homeostasis and inflammation. J Neuroinflammation. 2017; 14: 91. doi: 10.1186/s12974-017-0869-7
135. Le-Page-Devigry MT, Bidard JN, Rouvier F, Bulari C, Lazdunski M. Presence of abscicic acid, a phytohormonein the mammalian brain. Proc Natl Acad Sci U S A. 1986; 83: 1155-1158.
136. Minorsky PV. Abscicic acid, a universal signaling factor? Plant Physiol. 2007; 128: 788-789.
137. Gomez-Cadenas A, Vives V, Zandalinas SI, et al. Abscisic acid and a versatile phytohormone in plant signaling and beyond. Curr Prot Papt Sci. 2015; 16(5): 13-34. doi: 10.2174/1389203716666150330130102
138. Magnone M, Ameri P, Salis A, et al. Microgram amounts of abscicic acid in fruit extract improves glucose tolerance and reduces insulin secretionin rats and in human. FASEB J. 2015; 29: 4783-4793. doi: 10.1096/fj.15-277731
139. Fuce S, Barsile G, Bavestrello G, et al. Abscicic acid signaling through cyclic ADP-ribose in hydroid regeneration. J Biol Chem. 2004; 279: 39783-39788. doi: 10.1074/jbc.M405348200
140. Bassanganya-Rera J, Shoneckza J, Kingston DG, et al. Mechanism of action and medicinal application of abscicic acid. Curr Med Chem. 2010; 17: 457-478. doi: 10.2174/092986710790226110
141. Bruzzone S, Rodato N, Usai C, et al. Abscicic acid is an endogenous stimulator of insulin release from human pancreatic islets with cyclic ADP-ribose as second messenger. J Biol Chem. 2008; 283: 32188-32197. doi: 10.1074/jbc.M802603200
142. Gun AJ, Evans NP, Honterollas R, Bassanganya-Rera J. T cell PPAR γ is required for the anti-inflammatory effecicacy of abscicic acidagainst experimental IBD. J Nutr Biochem. 2011; 22: 812-819. doi: 10.1016/j.jnutbio.2010.06.011
143. Sturla L, Fresia C, Guida I, et al. LANCL2 is necessary forabscicic acid binding and signaling in human granulocytes and in rat insulinoma cells. J Biol Chem. 2009; 284: 28045-28057. doi: 10.1074/jbc.M109.035329
144. Lu P, Hontecillas R, Philipson CW, Bassanganya-Rera J. Lanthionine synthetase components C like protein 2 a new drug target forinflammatory diseases and diabetes. Curr Drug Targets. 2014; 15(6): 565-572. doi: 10.2174/1389450115666140313123714
145. Brenner JD, Shearer KD, McCaffery PJ. Retinoic acid and affective disorders:the evidence for an association. J Clin Psychiatry. 2012; 73: 37-50. doi: 10.4088/JCP.10r05993
146. Qi CC, Zhang Z, Fang H, et al. Antidepressant effects of abscicic acid mediated by the downregulation of corticotrophic realeasing hormone gene expression in rats. Int J Neuropsychopharmacol. 2014; 18(4): pyu006. doi: 10.1093/ijnp/pyu006
147. Gao Y, Ottaway N, Shriever SC, et al. Hormones and diet but not body weight controlypothalamic microglial circuitry.Glia. 2014; 62: 17-25. doi: 10.1002/glia.22580
148. Spielman LL, Little JP, Klegeris A. Inflammation and insulin/IGF1 resistance as the possible link between obesity and neurodegeneration. J Neuroimmuol. 2014; 273: 8-21. doi: 10.1016/j.jneuroim.2014.06.004
149. Sobesky JL, Barrientos RM, De May HS, et al. High fat diet consumption disrupts memory and primes elevations in hippocampal IL-1β an effect that can be prevented by dietary reversal or IL1 receptor antagonist. Brain Behav Immun. 2014; 20: 142: 22-32. doi: 10.1016/j.bbi.2014.06.017
150. Sanchez-Sarua S, Moustafa S, Garcia-A.viles A, et al. The effect of acscicic acid chronic treatment on neuroinflammatory markers and memory in a rat model of high fat diet induced neuroinflammation. Nutr Metab. 2016; 13: 73. doi: 10.1186/s12986-016-0137-3
151. Tseng AH, Barclay JL, Oster H. Interactions between endocrine and circadian systems. J of Mol Endocrinol. 2014; 52: R1-R6. doi: 10.1530/JME-13-0118
152. Bass J, Takahashi JS. Circadian integration and energetic. Science. 2010; 230: 1349-1354.
153. Ueda HR, Chon W, Adachi A, et al. A Transcription factor response element for gene expression during circadian night. Nature. 2002; 418: 534-539. doi: 10.1038/nature00906
154. Panda S, Antoch MP, Miller RH, et al. Coordinated transcription of key pathways in the mouse by circadian clock. Cell. 2002; 109: 307-320. doi: 10.1016/S0092-8674(02)00722-5
155. Golombek DA, Rosenstein RE. Physiology of circadian entrainment. Physiol Rev. 2010; 90: 1063-1102.
156. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: Organization and coordination of central and peripheral clocks. Annual Reviews of Physiology. 2010; 72: 517-549. doi: 10.1146/annurev-physiol-021909-135821
157. Vollmers C, Gill S, Di Tacchio I, Pulivarthy SR, Le HD, Panda S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc Natl Acad Sci U S A. 2009; 106: 21453-21458. doi: 10.1073/pnas.0909591106
158. Li Y, Sato Y, Yamaguchi N. Shift work and the risk of metabolic syndrome: A nested case control study. Int J Occup Environ Health. 2011; 17: 154-160. doi: 10.1179/107735211799030960
159. Karisson B, Knulsson A, Lindani B. Is there an association between shiftwork and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup Environ Med. 2001; 58: 747-752. doi: 10.1136/oem.58.11.747
160. Barclay JL, Husse J, Bode B, et al. Circadian desynchrony promotesmetabolic disruption in a mouse model of shift work. PLoS One. 2012; 7: e37150. doi: 10.1371/journal.pone.0037150
161. Foster RG, Kretzman L. The rhythms of life: What your body clock means to you! Exp Physiol. 2014; 99: 599-606. doi: 10.1113/expphysiol.2012.071118
162. Jones SG, Benca RM. Circadian disruption in psychiatric disorders. Sleep Medicine Clinics. 2015; 10: 481-493. doi: 10.1016/j.jsmc.2015.07.004
163. Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheraltissue circadian clock. Proc Natl Acad Sci U S A. 2008; 105: 15172-15177. doi: 10.1073/pnas.0806717105
164. Zhang D, Tong X, Arthurs B, et al. Liver clock protein BMAL1 promotes de novo lipogenesis through insulin Mtorc-AKT signaling. J Biol Chem. 2014; 289(37): 25925-25935; doi: 10.1074/jbc.M114.567628
165. Rey G, Cesbron F, Rougemont J, Reinke H, Brunner M, Naef F. Genome wide and phase specific DNA binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biology. 2011; 9: e1000595. doi: 10.1371/journal.pbio.1000595
166. Gachon F, Oiela FF, Schaad O, Descombes P, Scabier U. The circadian PAR- domain basic leucine zipper transcription factors DBP, TEF and HLF modulate basal and inducible xenobiotic detoxification. Cell Metb. 2006; 4: 25-36. doi: 10.1016/j.cmet.2006.04.015
167. De Boyne JP, Weaver DR, Dallman R. The hepatic circadian clock modulates xenobiotic metabolism in mice. J Biol Rhythms. 2014; 29: 277-287. doi: 10.1177/0748730414544740
168. Fuku K, Ferris HA, Kahn CR. Effect of cholesterol reduction on receptor signaling in neurons. J Biol Chem. 2015; 290: 26383-26392. doi: 10.1074/jbc.M115.664367
169. Grechez-Cassau A, Rayet B, Guillamond F, Teboui M, Delaumay F. The circadian clock component BMAL1 ia a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem. 2008; 283: 4535-4542. doi: 10.1074/jbc.M705576200
170. Turek FW, Joshu C, Kohsaka A, et al. Obesity and metabolic syndrome in circadian clock mutant mice. Science. 2005; 308: 1043-1045. doi: 10.1126/science.1108750
171. Arble DM, Sandova DA. CNS controlof glucose metabolism response to environmental challenges. Front Neurosci. 2013; 7: 20. doi: 10.3389/fnins.2013.00020
172. Rubic RD, McNamara P, Curtis AM, et al. BMAL1 and CLOCK-two established components of the circadian clock, are involved in glucose homeostasis. PLoS Biology. 2004; 2: e377. doi: 10.1371/journal.pbio.0020377
173. Barclay JL, Shostak A, Lelavski A, et al. High fat diet induced hyperinsulinemia and tissue specific insulin resistance in cry deficient mice. Am J Physiol Endocrinol Metab. 2013; 104: E1053-E1063. doi: 10.1152/ajpendo.00512.2012
174. Sherman H, Genzer Y, Cohen R, Channik N, Madat Z, Froy O. Timed high fat diet resets circadian metabolism and prevents obesity. FASEB J. 2012; 26: 3493-3502. doi: 10.1096/fj.12-208868
175. Cornejo MP, Hentges ST, Maliqueo M, Corini H, Racu-Wilacbos D, Elias CF. Neuroendocrine regulation of metabolism. J Neuroendocrinol. 2016; 28(7): doi: 10.1111/jne.12395
176. Waterson MJ, Horvath TL. Neuronal regulation of energy homeostasis. Cell Metab. 2015; 22: 962-970. doi: 10.1016/j.cmet.2015.09.026
177. Horvath TL, Diano S, Tschop M. Brain circuits regulating energy homeostasis. Neuroscientist. 2004; 10: 235-246. doi: 10.1016/j.regpep.2007.10.006
178. Xu J, Lloyd DJ, Hale C, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure and improves insulin sensitivity in diet induced obese mice. Diabetes. 2009; 58: 250-258. doi: 10.2337/db08-0392
179. Dhawan R, Fellet C, Costa SS, et al. Liver derived ketone bodies are necessary for food anticipation. Nat Commun. 2016; 7: 10580. doi: 10.1038/ncomms10580
180. Schwartz MW, Woods SC, Porte Jr C, Seeley BJ, Baskin DG. Central nervous system control of food intake. Nature. 2000; 404: 661-671. doi: 10.1038/35007534
181. Yi CX, Tschop MH. Brain-gut-adipose tissue communication pathways at a glance. Dis Model Mech. 2012; 5: 583-587. doi: 10.1242/dmm.009902
182. Meyer-Kovac J, Kolbe I, Ehrhardt L, et al. Hepatic gene therapy rescues high fat diet responses in circadian Clock mutant mice. Molecular Metab. 2017; 6: 512-523. doi: 10.1016/j.molmet.2017.03.008
183. Li C, Yang J, Yu S, et al. Triterperiod sapponins with neuroprotective effects from the roots of Polygala Tenuifolia. Planta Med. 2008; 74(2): 133-141.
184. Jiang Y, Zhang W, Tu F, Xu X. Polygala Tenuifolia. And their conformational analysis. J Nat Prod. 2005; 68(6): 875-879.
185. Le TK, Jeong JJ, Kim DH. Clionosterol and ethyl cholestan-22-enolisolated from the rhizome of Polygala Tenuifolia inhibit phosphatidylinositol-3kinase’/Akt pathway. Mol Pharm Bull. 2012; 35(8): 1379-1383. doi: 10.1248/bpb.b12-00426
186. Pari L, Tewas D, Eckel J. Role of curcumin in health and disease. Arch Physiol Biochem. 2008; 114(2): 127-149. doi: 10.1080/13813450802033958
187. Arun N, Nalini N. Efficacy of turmeric on blood sugar and polyol pathway indiabetic albino rats. Plants Foods Hum Nutr. 2002; 57(1): 41-52. doi: 10.1023/A:1013106527829
188. Ramadan G, Al-Kahtani M, El Sayed WM. Antiinflammatory ans antioxidant properties of Curcuma longa (turmeric) versus Zingiber officinale (ginger) rhizomes in rat adjuvant induced arthritis. Inflammation. 2011; 34(4): 291-301. doi: 10.1007/s10753-010-9278-0
189. YuWF, Kwan PL, Wong CY, et al. Attenuation of fatty liverand prevention of hypercholesterolemia by extreact of curcumina longa through regulating the expression of CYP7A1, LDL-receptor HO-1 and HMG-COA reductase. J Food Sci. 2011; 76(3): 1180-1189. doi: 10.1111/j.1750-3841.2011.02042.x
190. Chung BS, Chin MG. Dictionary of Korean Folk Medicine. Incheon, South Korea: Young Lim Publishing Co Ltd; 1990: 813-914.
191. Wang ES, Zhao DQ, Cheng DY, Liu YH. Ent-Sauchinone from Saurus chinensis. Heterocytes. 2008; 75(5): 1241-1246.
192. Yu MH, Im HG, Lee JW, et al. Effects of ehanol extract from Saurus chinensis (Bour) Baill on lipid and antioxidantmetabolism in rats fed a high fat diet. Nat Prod Res. 2008; 22(3): 275-283. doi: 10.1080/14786410701590657
193. Yun YR, Kim MJ, Kwon MJ, Kim HA, Song KB, Song YO. Lipid lowering effect of hot water soluble extracts of Saurus chinensis Bail on rats fed high fat diet. J Med Food. 2007; 10(2): 316-322. doi: 10.1089/jmf.2006.149
194. Joo HJ, Kang MJ, Seo TJ, et al. The hypoglycaemic effect of Saurus chinensis Bail in animal models of diabetes mellitus.Food Sci Biotechnol. 2006; 15(3): 413-417. doi: 10.1017/S0022029914000582
195. Kim SR, Sung SH, Kang SY, et al. Aristolactam Bill OF Saurus chinensis attenuates glutamate induced neurotoxicityin rat cortical cultures probably by inhibiting nitric oxide production. Plant Med. 2004; 70(5): 391-396.
196. Sung SH, Kim YC. Hepatoprotective diasteromeric lignanas from Saurus chinensis herbs. J Natl Prod. 2000; 63(7): 1019-1021. doi: 10.1021/np990499e
197. Zhu X, Jing I, Chen C, et al. Danzhi Xiaoyao San amelioratesdepressive–like behavior by shifting toward serotonin via the downregulation of hippocampal indoleamin e2,3 dioxygenase. J Ethnopharmacol. 2015; 160: 86-93. doi: 10.1016/j.jep.2014.11.031
198. Oh HI, Park HB, Ju MS. Comparitive study of antioxidant and antiinflammatoryactivities between curcumin longae radix and curcumin longae rhizona. Korea J Herbology. 2010; 25(1): 83-91.
199. Roh JS, Lee H, Woo S, et al. Herbal composition Gambigyeeongsihwan (4) from curcumin longa Alnusjaponica, and Massa Medicata fermentata inhibits lipid accumulation in 3T3L1 cells and regulates obesity in Otsuka long Evans Tokushima fatty rats. J Ethno Pharmacol. 2015; 171: 287-294. doi: 10.1016/j.jep.2015.05.056
200. Has JM, Lee JS, Kim HG, et al. Synergistic effects Artermisia Imayomogi and curcumin longae radix on high fat dietinduced hyperlipidemia in a mouse model. J Ethno pharmacol. 2015; 173: 217-224. doi: 10.1016/j.jep.2015.07.021
201. Huang KC. The Pharmacology of Chinese Herbs. Boca Ratnon, FL, USA: CRC Press; 1991.
202. College JNM. Dictionary of Chinese Drugs Shanghai. HongKong Island, Hong Kong: Shanghai Scientific Technologic Publisher; 1997
203. Chen YL, Hsieh CL, Wu PH, Lin JC. Effect of polygala tenuifolia on behavioral disorders of lesioning nucleus basalis magnocellularis in rat. J Ethno pharmacol. 2004; 95(1): 47-55. doi: 10.1016/j.jep.2004.06.015
204. Spelman K, Burns J, Nichols D, Winters N, Offersberg N, Fenninnorg M. Modulation of cytokine expression by traditional medicines: A review of herbal immunomoodulators. Alter Med Rev. 2006; 11(2): 128-150. doi: 10.1016/j.jep.2015.07.021
205. Lee JH, Lee JJ, Cho WK, et al. KBH1, an herbal composition, improves hepatic steatosis and leptin resistance in high fat diet induced obese mice. BMC Complement Altern Med. 2016; 16: 355. doi: 10.1186/s12906-016-1265-z
206. Milanski M, Degasperi G, Coope A, et al. Satturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamic implications for the pathogenesis of obesity. J Neurosci. 2004; 29(2): 359-370. doi: 10.1523/JNEUROSCI.2760-08.2009
207. Schwartz MW, Porte D Jr. Diabetes, obesity and the brain.Science. 2005; 21: 307(5708): 375-379. doi: 10.1126/science.
1104344
208. Williams KW, Elmquist JK. From neuroanatomy to behavior: Central integration of peripheral signals regulating feeding behavior. Nat Neurosci. 2012; 15(10): 1350-1355. doi: 10.1038/nn.3217
209. Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2011; 108: 2939-2944. doi: 10.1073/pnas.1006875108
210. Kleinridders A, Schenlen D, Konner AC, et al. MyD88 signaling in the CNS is required for development of fatty acid induced leptin resistance and diet induced obesity. Cell. 2009; 10: 249-259. doi: 10.1016/j.cmet.2009.08.013
211. Belgardt BF, Mauer J, Wunderlich FT, et al. Hypothalamic and pituitary c-jun N terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Nat Acad Sci U S A. 2010; 107: 6028-6033. doi: 10.1073/pnas.1001796107
212. Sabio G, Cavanagh-Kyros J, Barett T, et al. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulationby JNK1. Genes Dev. 2010; 24: 256-264. doi: 10.1101/gad.1878510
213. Unger EK, Piper ML, Olofsson LE, Xu AW. Functional role of c-jun-N terminal kinase in feeding regulation. Endocrinology. 2010; 151: 671-682. doi: 10.1210/en.2009-0711
214. Moraes JC, Coope A, Morari J, et al. High fat diet induces apoptosis of hypothalamic neurons. PLoS One. 2009; 4(4): 5045. doi: 10.1371/journal.pone.0005045
215. McNay DE, Briancon N, Kokoeva MV, Maralos-Flier E, Flier JS. Remodeling of the arcuate nucleus o energy balance circuit is inhibited in obese mice. J Clin Invest. 2012; 122(1): 153-162. doi: 10.1172/JCI43134
216. Van de Sande-Lee S, Pereira FR, Cintra DE, et al. Partial reversibility of hypothalamic dysfunction and changes in brain activity after body mass reduction in obese subjects. Diabetes. 2011; 60(6): 1699-1704. doi: 10.2337/db10-1614
217. Li J, Tang Y, Cai D. IKK beta/NF kappa B disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and prediabetes. Nat Cell Biol. 2012; 14: 999-1012. doi: 10.1038/ncb2562
218. Steinberg GR, Kemp BE. AMPK in healthand disease. Physiol Rev. 2009; 89: 1025-1078. doi: 10.1152/physrev.00011.2008
219. Kalsbeek A, Bruinstroop E, Yi CX, Khoverick LP, La-Fleur SE, Fliers E. Hypothalamic control of energy metabolism via the autonomic nevous system. Ann NY Acad Sci. 2010; 1212: 114-129. doi: 10.1111/j.1749-6632.2010.05800.x
220. Mong Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the I kappa B kinase beta(IKKbeta)/NF-kappa B pathway. J Biol Chem. 2011; 286(37): 32324-32332. doi: 10.1074/jbc.M111.254417
221. Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010; 90(4): 1383-1435. doi: 10.1152/physrev.00030.2009
222. Yang Z, Kilonsky D. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009; 335: 1-32. doi: 10.1007/978-3-642-00302-8_1
223. Glick D, Barth S, Macleod KF. Autophagy, cellular and molecular mechanisms. J Pathol. 2010; 221(1): 3-12. doi: 10.1002/path.2697
224. Shi CS, Shendrov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1β Production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012; 13(3): 255-263. doi: 10.1038/ni.2215
225. Harris J, Hartman M, Roche C, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011; 286(11): 9587-9597. doi: 10.1074/jbc.m110.202911
226. 226. Portovedo M, Ignacio-Souza LM, Bombassaro B, et al. Saturated fatty acids modulate autophagy’s proteins in the hypothalamus. PLoS One. 2015; 10(3): e0119850. doi: 10.1371/journal.pone.0119850