INTRODUCTION
Numerous reports have described the mechanisms for cocaine (COC) and methamphetamine (MA) action in the central nervous system (CNS) over the last 3-4 decades. For both COC and MA, it is clear that dopamine is an important component in the addiction, euphoria and dependency processes.1,2,3 The extracellular chemical environment of the nerve synapse may alter drug responsiveness and therein influence risk factors associated with drug taking. The mechanisms of heavy metal action on the CNS is less clear, plagued by the inability of some metals to cross the blood-brain barrier. Our understanding is increasing, and in vivo studies have provided the foundation for most of our understanding.4,5,6 The effects of lead (Pb) on dopaminergic function have been well described.7,8,9 Pb is known to target the mesolimbic dopamine (DA) system, primarily projection neurons from the ventral tegmental area to the nucleus accumbens.7 Since DA activity along this circuit is critically involved in determining COC responsiveness,10 functional disturbances in mesolimbic DA operations resulting from perinatal or postnatal Pb exposure may translate into persistently increased sensitivity to COC. Both Pb and COC affect the release and uptake of DA.8,9,11,12 There was a need to pursue other metal/drug interactions, such as mercury (Hg) and MA, which are known to affect the DAT.13,14,15,16 Prior work has also suggested that Hg has achieved access to striatal tissue,17 due to the increased levels of DA detected when exposed to mercury (HgCl2).14 Previous work has shown that Hg blocks the degradation pathway for catecholamines,18 including DA. A large body of evidence examining the effects of MA on the dopaminergic system has demonstrated long-lasting decreases in DA levels, tyrosine hydroxylase activity, and DA uptake sites in the brain.19,20,21,22 Many studies have demonstrated modifications in the DAT function, which may be associated with either/both the altered uptake and release of DA.19,20,21,22,23,24,25 The long-acting changes observed following MA exposure can be due to the destruction of DA neurons. MA has been shown to be a neurotoxin following exposure to higher concentrations or doses, for longer durations.20,22,23 The belief that mixtures of toxicants/toxins can elicit additive or synergistic toxicity has led to a heightened interest in the toxicological studies of these mixtures.
Both COC and MA are psychostimulants classified as Schedule II drugs (C-II) in the United States. This schedule represents drugs, which still have medicinal use, but exhibit a high degree of addiction/dependence/health hazards. COC can be snorted, smoked or injected and the route of administration will dictate the intensity of the ‘high’ and the duration that the high will last. Being a stimulant, (ab)use of COC will lead to increased physiological responses, such as increased heart rate, increased blood pressure, anxiety, paranoia, and irritability.26 Although, the exact mechanism differs with MA action in the CNS, the generalized symptomology is similar between COC and MA.27 In the past, an oversimplified view of psychostimulant action has been that COC blocks DA uptake, leading to increased DA transmission and the physiological and behavioral changes observed following COC administration. Not only does COC reduce the uptake of DA both in vivo and in vitro,2,28,29 but COC administration also results in increased DA in the synapse through non-antagonist mechanisms.3,30 Increased synaptic DA is thought to be due to a reverse transport of DA through the DAT.3In vivo analysis indicates that elevated synaptic DA is not due to increased firing rates of dopaminergic neurons since neither frequency nor amplitude of DA release is changed.2 COC interference with DA uptake is believed to be due to a competitive interaction with DA in the binding pocket on DAT.29,31 Changes in vivo following COC administration are characterized by a reduction in DA uptake due to a reduction in Km, or affinity, of DA for the DAT which occurred after a long exposure to COC.28 Immediate effects on DAT indicate that an increased clearance occurs in response to elevated extracellular DA, partially explaining the ability of DAT inhibitors to increase DA clearance rates.32 In human abusers of COC, imaging analysis revealed an increase in the number of DAT molecules on the cell surface, most likely an adaptive measure to elevated synaptic DA.11 Molecular studies have substantiated in vivo reports demonstrating increased expression of DAT mRNA, increased production of DAT protein and increased trafficking or movement to the cell surface.29,30,33,34 Contrary to other findings, Daws et al12 reported an increase in DA uptake following exposure to COC. They concluded that after exposure and washing the COC out of the culture, COC had triggered an increase in DAT insertion into the cell membrane. By removing the COC before assessing DAT function, the ability of inhibitory action of COC on DA binding to DAT is eliminated. Combining neurochemical and molecular studies with the behavioral outcomes after COC administration suggests that COC is acting through additional neural systems and not just DA.2,29
The action of MA on the dopaminergic system is more complex than COC. In primary striatal cultures, MA exposure reduces the Km of DA for DAT without changes in either Vmax or Bmax.23 Changes in DAT kinetics following MA exposure demonstrate a regional variation in the striatum displaying greater sensitivity to the actions of MA.22,24 Fleckenstein et al15,35 compared the actions of COC and MA following single or multiple injections. The IC50 values for COC (337 nM) were marginally different compared to MA (291 nM), but the effects on DA uptake where robust. COC only slightly altered DA uptake after a single injection whereas MA reduced dopamine uptake to 63% of control values after a single injection and 30% of control after multiple injections. The authors speculated that MA alters DA release and uptake through multiple mechanisms such as reverse transport, exchange diffusion, phosphorylation changes, and alterations in protein trafficking. Previous reports state COC increases the trafficking of DAT to the cell surface, exposure to MA results in the internalization of DAT, which leads to a reduction in DA uptake.34,36 It is clear that the actions of COC in the addictive process and its actions via DAT are much more complex than previously believed and could be under multiple influences from other neurotransmitter systems.
The role of heavy metal-mediated changes in CNS receptor or transporter function is not as well understood as with COC and MA. The earliest reports suggest that Pb, Hg and other divalent metals can interact with binding sites on muscarinic receptors in a competitive fashion.37,38 Many reports describe the behavioral and learning changes following exposure to Pb7,39,40,41 or Hg4,18,42, but have been unable to specify a particular neurotransmitter system. Behavioral effects after Pb exposure are dependent on the neurotransmitter system affected.7 How the DA system is impaired is still unclear. Exposure to Pb is reported to increase messenger ribonucleic acid (mRNA) for both tyrosine hydroxylase and DAT,6 but in vivo administration of Pb results in a decrease in DA uptake.9 Seemingly conflicting, these data can be explained that more mRNA is present to adapt for the reduction in DA uptake. Additional work is needed to determine if the Pb-mediated increase in DAT mRNA translates to an increase in DAT protein production and trafficking to the cell membrane. Contrary to the Pb-mediated effect of DA hypofunction, the Hg-mediated effects suggest DA hyperfunction. Faro et al17,43,44,45,46 has extensively studied the effects of Hg (both organic and inorganic) on the DA system and has universally reported a significant increase in DA release. They suggest three mechanisms by which synaptic DA is increased a) increased DA release, b) inhibition of DA metabolism and c) inhibition of DA uptake. Collectively, the mechanisms for organic and inorganic Hg effects are different.46 Organic Hg both increases DA release and inhibits uptake, yielding a net increase in synaptic DA. One mechanism by which Hg can alter uptake is by effecting the kinetic profile of the DAT. The presence of Hg ions in a binding buffer increased the binding of both [3H] WIN35,428 and [3H] mazindol to DAT.13 This increase in binding could be due to either increase in the number of DAT or the affinity of DAT for the radioligands. The ability of inorganic Hg to alter [3H] methylphenidate binding to DAT exhibited a biphasic response.47 The initial phase, increased binding, represents a rapid increase in affinity (Kd), which is followed by a reduction in binding due to a reduction in both affinity (Kd) and density (Bmax). This biphasic response suggests that the timing of analysis is paramount and that a thorough examination of a time-course would be necessary. A more recent study suggests that a more complex model exists for the actions of first-row transition metals with DAT resulting in a reduced affinity of DAT for DA.48 Pb is a post-transition metal, and Hg is one of the third-row transition metals, so this reduction in affinity may not apply to Pb and Hg but offers an interesting perspective on potential metal-mediated effects on DAT. Similar to observations with COC and MA, it is obvious that Pb and Hg are mechanistically different regarding their actions on the DA system. This complexity and involvement of other neurotransmitter systems, such as glutamate, increases the possibility of interactions between heavy metals and psychostimulants.
As our understanding of metal-induced toxicity has increased, humans have tried to reduce their exposure. Immediate reductions in exposure have not addressed the ability of these metals to persist in the environment for extended periods have led to a continuing health concern. Although the use of psychostimulants such as COC and MA have significantly declined over the last twenty years, their use has plateaued and maintained a constant level of consumption. Although the mechanism is complex, the actions of COC and MA in the CNS involve enhancement of DA transmission.1 The possibility exists that exposure to heavy metals may alter DAT function in a manner such that use of COC or MA result in an amplified response and increase the abuse potential of these drugs.5 The purpose of this study was to gain insight into the interaction of heavy metals and drugs of abuse at DAT. The hypothesis of the present study is exposure to individual agents (or combinations) will significantly alter DAT function as indicated in binding and uptake studies. We did intend to examine the effects of Pb, Hg, COC, and MA on DAT function in N2A cells. By examining different time point and utilizing mixtures of these agents, we will develop a better understanding of the actions of heavy metals and psychostimulants on DAT with implications on human health. Also, the intensity of psychostimulant response may be predicated on prior exposure to environmental toxicants such as heavy metals.
MATERIALS AND METHODS
Cell Culture
The cell line used was the Neuro2A (N2A; mouse neuroblastoma cells), provided by American Type Culture Collection (CCL-131; ATCC, Manassas, VA, USA). Upon arrival, cells were rapidly thawed and added to pre-warmed growth media, Minimum Eagles Media (MEM) with Earle’s balanced salt solution, nonessential amino acids, and sodium pyruvate. Media was supplemented with: 2 mM L-glutamine, 1.5 g/L NaHCO3, 10% heat-inactivated fetal bovine serum (FBS), and 1% penicillin/streptomycin (10,000 I.U./mL and 10,000 µg/mL, respectively; Cellgro (MediaTech Inc., Herndon, VA, USA and Hyclone, Logan, UT, USA). Cultures were maintained in 25 cm² cell culture flasks with vented caps (Corning Inc., Corning, NY, USA) at 37 °C in a 5% CO2 humidified atmosphere. Culture medium was exchanged every other day. When cells were close to confluence (usually within one week), trypsin (0.25%) was added to the flask and once the cells were detached, aliquots were transferred to fresh flasks with media. N2A cells were subcultured at a ratio of 1:4 to 1:6 depending on their growth.
For cell viability, binding and uptake assays, 1×105 cells were added to each well of a 24-well plastic cell culture plate (Costar® 3599, Corning, Inc., Corning, NY, USA). Plates were returned to the incubator (37°C/5% CO2) and the cells were allowed to continue their normal growth until assayed. Cells were normally >80% confluence at the time of assay and only cells within the passage range of 4-14 were used for determining the effects of heavy metal/psychostimulant effects.
Transfection of N2A Neuroblastoma Cells with hDAT
Vectors, pCMV6-XL5 with the hDAT-cDNA insert (Origene) and pCMV6-Neo (Origene), were transformed into One Shot® Top10 Chemically Competent E. coli (Invitrogen). Cells were grown on LB-agar/ampicillin (100 µg/mL) plates overnight. The following day six independent colonies were selected to inoculate 2 mL LB/ampicillin broth overnight. Plasmid DNA was isolated using the Wizard® Plus SV Minipreps DNA Purification System kit (Promega). Both plasmids were digested with Not I endonuclease (Promega). Fragments were separated by 0.7% agarose gel electrophoresis, and extracted using the StrataPrep® DNA Gel Extraction kit (Stratagene). DNA was quantified by spectrophotometric analysis. The hDAT-cDNA and the pCMV6-Neo were ligated with T4 DNA ligase (Promega) at 4 ºC overnight. The new subcloned construct was transformed into Top10 cells and grown again on LB-agar/ampicillin plates. LB/ampicillin broth was inoculated with selected colonies, and the resulting pDNA was isolated by Miniprep. Correct insertion was confirmed following XmaI digestion and DNA sequencing. Confirmed plasmids were plated for colonies, inoculated in 100 mL LB/ampicillin broth, and purified using the Qiagen Plasmid Maxi kit. The cloned plasmid pCMV6-Neo-(hDAT) was quantified by spectrophotometric analysis, and aliquots were frozen back for use in assays.
One day before transfection, cells were subcultured into a 24-well plate at a density of 1×105 cells per well in 500 μL of growth medium without antibiotics. Confluence was 90-95% at the time of transfection. Cells were transfected with Lipofectamine™ 2000 (Invitrogen) according to manufacturer’s instructions. The transfection mixture was comprised of; culture medium (500 μL), plasmid DNA (0.8 μg), Lipofectamine™ 2000 (2.0 μL) and Opti-MEM I® (Invitrogen) as a diluent. Lipofectamine™ 2000 is a cationic liposome formulation that functions by complexing with nucleic acid molecules, overcoming the electrostatic repulsion of the cell membrane and allowing entrance of the nucleic acid. Media was changed after 4-6 h and cells were incubated at 37°C in a 5% CO2 incubator for 24 h prior to testing for transgene expression. For stable transfection, cells were passed 1:10 into fresh growth medium 24 h after transfection. The following day Geneticin was added to the culture medium to yield a final concentration of 500 µg/mL. Transfected cell selection continued for three weeks.
Tagging hDAT with Green Fluorescent Protein (GFP)
A fluorescently tagged hDAT was constructed by fusing the N terminus-encoding region of the enhanced green fluorescent protein (eGFP) cDNA from pEGFP-N3 (BD Biosciences Clontech, Palo Alto, CA, USA) to the C terminus encoding region of the human synthetic DAT cDNA from pCMV6-XL5 (Origene, Rockville, MD, USA), thereby creating the fusion construct hDAT-GFP. This construct was subcloned into a pIRES-Neo3 expression vector modified to express the synthetic hDAT from a cytomegalovirus promoter and a neomycin resistance gene. Neuro2A (N2A) cells were transfected with the hDAT-GFP using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA). Correct insertion was confirmed following KpnI digestion and DNA sequencing. A stably transfected pool (hDAT cells) was selected in 500 µg/mL geneticin (Invitrogen, Carlsbad, CA, USA). Cells were cultured in supplemented MEM as described in the ‘Cell Culture’ section, except that cells were grown in 75 cm² cell culture flasks with vented caps (Corning Inc., Corning, NY, USA) at 37°C in a 5% CO2 humidified atmosphere. N2A cells containing the hDAT-GFP construct were grown in normal N2A growth media to confluence, and then plated at a density of 1×105 cells/well in a 24-well plate. Once in the plate, cells were grown to at least 80% confluence before assaying.
Cell Viability – Lactate Dehydrogenase Assay
We quantified released LDH using a colorimetric assay measuring LDH activity in the media (CytoTox 96® Non-Radioactive Cytotoxicity Assay; Promega). Briefly, released LDH activity in culture media using a 30-minute coupled enzymatic assay, measuring the amount of a red formazan product. The measured absorbance is directly proportional to the number of lysed cells. The viability of cells in the treatment groups [0.1, 1, or 10 µM HgCl2 or PbCl2, and 10 or 100 nM psychostimulants (COC; MA) for 24, 48, 72, or 96 h (Promega; Madison, WI, USA) were compared to values obtained from the vehicle control groups. 50 µL of each sample supernatant was transferred in duplicate into a clear 96-well plate to quantify spontaneous LDH release (control). The remaining media from the 24-well plate was aspirated. To each well of the 24-well plate, 150 µL of lysis solution (0.9% Triton-X) was added to measure the total amount of LDH in each well. Plates were returned to the 37ºC incubator for 45 minutes. Afterwards, 900 µL of complete media was added to each well to bring the total volume back up to the original volume of 1050 µL. Again, 50 µL of each sample supernatant was transferred in duplicate into the 96-well plate. Next, 50 µL of activated substrate mix (Assay buffer = Tris-buffered tetrazolium dye, Triton-X-100, lyophilized diaphorase, lactate, and NAD+) was added to each of the 96 wells protecting from light by covering with foil. Covered plates were placed on a plate shaker for 30 min and the reaction was stopped by the addition of 50 µL of stop reagent (1M acetic acid). Absorbance was measured using a Synergy HT microplate reader (Bio-TEK® Instruments, Inc., Winooski, VT, USA) at 490 nm. Cell viability was determined by the following equation:
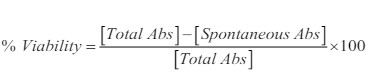
[3H]GBR-12,935 Binding
N2A (hDAT) cells were washed with 1 mL of assay buffer (50 mM Tris-HCl and 120 mM NaCl; pH 7.7), followed by addition of 200 μL trypsin/EDTA. The resulting cell suspension is centrifuged at 3,000 x g for 5 min. Pelleted cells were resuspended in 1.25 mL assay buffer and 400 μL of this cell suspension was transferred into binding tubes with either 50 μL of assay buffer (total binding) or 50 μL of 50 μM GBR12909 (non-specific binding; 5 μM, final concentration). To initiate the binding reaction, 50 μL of 500 nM [3H] GBR12935 was added to all tubes (43.0 Ci/mmol; Perkin Elmer, UK; 50 nM, final concentration). All samples were run in duplicate. Tubes were vortexed followed by a 60 min incubation period at room temperature (21-22 °C) to reach equilibrium. The binding reaction was terminated by filtration under reduced pressure using a tissue harvester (Brandel, Gaithersburg, MD, USA) onto Whatman GF/C filters that had been pre-soaked in 0.3% polyethyleneimine (PEI). Filters were washed with ice-cold 0.9% NaCl for 15 sec and then placed in scintillation vials with 5 mL of ScintiVerse BD cocktail (Fisher Scientific, Pittsburgh PA, USA). To determine the quantity of [3H] bound, the number of CPM was determined by scintillation spectrophotometry based on the number of DPM counted in a 5 minute period and factoring the efficient of the scintillation counter (Beckman Coulter LS 1801; Fullerton, CA, USA).
[3H]Dopamine Uptake
Clearance of synaptic dopamine is the responsibility of the DAT and is important for the maintenance of neurotransmitter homeostasis. N2A-hDAT cells were grown as previously described and plated prior to the start of the assay. Once cells reached >80% confluence, each well was washed with 1 mL Krebs-HEPES uptake buffer (mM) [25 HEPES, 120NaCl, 5 KCl, 2.5 CaCl2, 1.2 MgSO4, 0.3 ascorbic acid, 0.001 pargyline and 2 mg/mL D-(+)-glucose; pH 7.4]. After washing, cells were detached by the addition of 200 μL trypsin/EDTA. Each aliquot of cell suspension was centrifuged at 3,000 x g for 5 min. Resulting pellets were resuspended in 1.25 mL Krebs-HEPES uptake buffer and 400 μL was transferred into 12×75 mm polypropylene tubes. Either 50 μL of uptake buffer (total binding) or 50 μL of 50 μM GBR12909 (non-specific uptake; 5.0 μM, final concentration) was added to parallel tubes. Uptake was initiated by the addition of 50 μL [3H] DA (48.0 Ci/mmol; Amersham Biosciences, UK; 100 nM, final concentration) to all tubes. The tubes were vortexed followed by 10 min incubation at room temperature, then terminated by filtration onto a Whatman GF/C filter that had been pre-soaked in 0.3% PEI. Following filtration, filters are washed with ice-cold 0.9% NaCl for 15 sec and then placed in vials with 5 mL of scintillation cocktail. To quantify the uptake of [3H] DA into the cells, the number of CPM was determined by scintillation spectrophotometry based on the number of DPM counted in a 5 minute period and factoring the efficient of the scintillation counter (Beckman Coulter LS 1801; Fullerton, CA, USA).
Statistical Analysis
All data that expressed represent the group means±standard error of the mean (SEM). For LDH assays, collected data was absorbance @ 490 nm and then using the formula described above, converted to percent viability. For viability studies, one-way ANOVA was performed on the raw, non-transformed data followed by Dunnett’s posthoc test for comparison to control. We assigned the vehicle-24 h group for each metal as its control value. From these data, we can determine what concentrations and exposure times would be appropriate in subsequent studies such that the density or function of hDAT would not be compromised due to cellular damage or death. Data collected from the binding of [3H] GBR12935 and uptake of [3H] dopamine was first normalized to protein content. Specific binding (and uptake) was calculated as the difference between binding (and uptake) in the absence and presence of the specific binding (or uptake) inhibitor of DAT. Specific CPM/mg protein was then divided by specific activity of the radioligand. Results were expressed as fmol/mg protein for binding and nmol/mg/min for uptake. [3H]GBR12935 binding assays and [3H] dopamine uptake assays involving one treatment time with multiple treatments were analyzed using two-way analysis of variance (ANOVA) (metal x stimulant), followed by Sidak’s posthoc multiple comparisons. All computer analyses of data were performed using Prism v7 (GraphPad Software 7.04; San Diego, CA, USA). The level of statistical significance was set at α=0.05.
RESULTS
Transfection of N2A Cells with hDAT and hDAT-GFP
Development of transient clones using the pCMV6-XL5 vector was successful as indicated by the production of 5.8 kb and 2.7 kb bands following digestion with NotI (Figure 1A and B). We desired to generate a stable clone in addition to the transient expression observed in Figure 1B, so hDAT was subcloned into a pCMV6-Neo vector (Figure 2A). The presence of the neomycin resistant gene allowed for the selection of stable clone over time, which were cryopreserved for later use. Plasmids were digested with XmaI yielding fragments of 4.8 kb, 2.7 kb, and 1 kb (Figure 2B) suggesting a correctly oriented clone. N2A cell stably expressing hDAT were grown in 75 cm2 flasks and 1 mL aliquots were cryopreserved in cryopreservation media of base minimum essential medium (MEM) with 5% dimethyl sulfoxide (DMSO) (approximately 1 mg of cells/mL). Aliquots were stored in the vapor phase of a liquid nitrogen cryopreservation Dewar.
Figure 1. The pCMV6-XL5 Vector (~4.5 kb) with the hDAT cDNA (about 2.7 kb) Inserted within the Multiple Cloning Site (A). The hDAT Insert is Flanked by Two Not I Restriction Sites. ColE1 is the Bacterial Origin of Replication and SV40 Allows forReplication in Mammalian Cells. We used the CMV Promoter to Express the Cloned cDNA. Plasmid Selection in E. coli is Facilitated by the Expression of the Ampicillinresistance Gene. Plasmids were Digested with Not I and Fragments Separated by 0.7% Agarose Gel Electrophoresis. GeneMate QuantiMarker 1 kb was Used as the DNA Ladder. Lanes 1, 5, 6, 7, and 8 Show Fragments 5.8 kb and 2.7 kb in Size, Respectively (B). These are Successful pCMV6-Neo with the Cloned Insert.
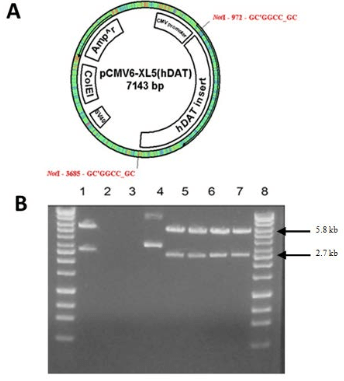
Figure 2. The pCMV6-Neo Vector (~5.8 kb) is Similar to pCMV6-XL5 Except for the Addition of a Neomycin Resistance Gene for Establishing a Stable Clone [A]. The hDAT cDNA was Subcloned into the Vector by Not I Digestion and Ligation. Orientation of the Insert was Confirmed by Xma I Digestion. The Cloned Plasmid was Transfected into COS-7 Cells. Successfully Cloned Plasmids were Digested with Xma I. Fragments were Separated by 0.7% Agarose Gel Electrophoresis. GeneMate Quanti-Marker 1 kb was Used as the DNA Ladder. Lanes 1, 3, 4, 5, and 6 show Fragments 4.8 kb, 2.7 kb, and 1.0 kb in Size, Respectively [B]. These are pCMV6-Neo with the Correctly Oriented Cloned Insert. Lane 7 is the pCMV6-Neo (5.8 kb) without Insert.
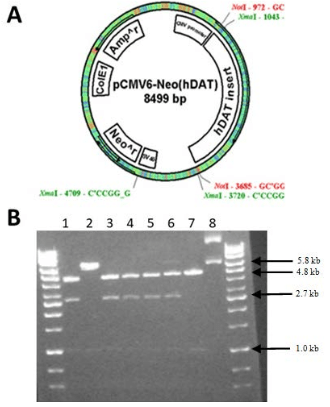
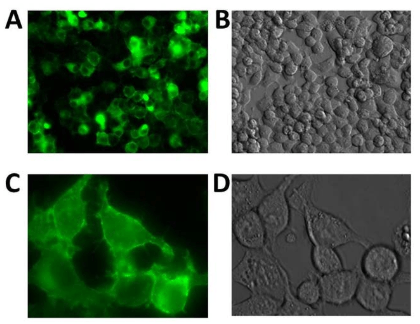
Using the hDAT-GFP fusion permitted the visualization of hDAT in N2A cells using fluorescent microscopy and allowed us to observe the localization of hDAT-GFP within N2A cells and determine the intracellular localization of hDAT. The vector, pIRES-N3 (Figure 3A) was used with hDAT-GFP inserted and flanked by EcoRI and NotI restriction sites. The plasmid was digested using KpnI and fragments of 4.4, 2.0 and 1.5 kb were generated (Figure 3B) suggesting a correctly cloned insert. N2A cells stably expressing hDAT-GFP were cryopreserved as described above. We visualized the expression of hDAT-GFP (Figure 4A and B) and found that hDAT was expressed on the cell membrane in a delimited fashion. There were distinct clusters, punctate, formations on the cell membrane representing hDAT localization, with only minimal aggregation in intracellular organelles (Figure 4C).
Figure 3. pIRES-N3 Vector (About 5.2 kb in size) with hDAT-GFP (About 2.6 kb) Inserted into the Multiple Cloning Site. The hDAT-GFP Insert isFlanked by Eco RI and Not I Restriction Sites [A]. Plasmids were Digested with Kpn I. Fragments were Separated by 1.0% Agarose Gel Electrophoresis. GeneMate QuantiMarker 1 kb was Used as the DNA Ladder. Lane 2 is an Empty pIRES-N3 Vector. Lanes 3 through 8 Indicate Fragments of the Approximate Size 4.4, 2.0, and 1.5 kb [B]. These are Correctly Cloned pIRES-N3 (hDAT-eGFP)
Figure 4. Epi-Fluorescent Photomicrographs of N2A Cells Transfected with GFP-Tagged hDAT. Panels [A] and [B]: 20x Images Used to Demonstrate Transfection Efficiency. Transfection Efficiency is Near 90% Using this Procedure. Panels [C] and [D]: 40x Localization Images of GFP-Tagged hDAT. These Images Suggest that the Tagged hDAT is Expressed on the Membrane in a Delimited Manner, but will Examine this Finding by Performing [3H] Dopamine Uptake and [3H] GBR12935 Binding. It does not Appear that Tagged hDAT is Aggregating or Retained in the Endoplasmic Reticulum. Photos were Obtained using an Olympus IX71 Epi-fluorescence Microscope Fitted with a FITC Filter and CoolSnapMonochrome Camera. The Pictures were Obtained and Colored Green Using Rsimage (Roper Imaging) Version 1.7.3.
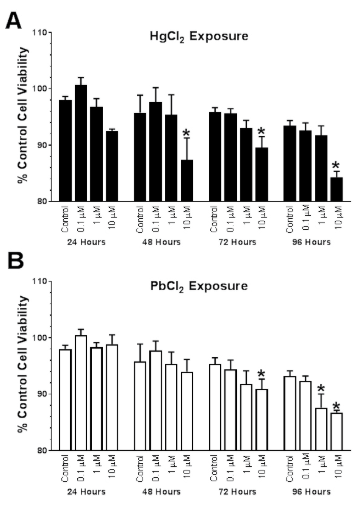
Cell Viability Following Metal Exposure: Time Course and Concentration Curve
Lactate dehydrogenase (LDH) activity was measured at four-time points between 24-96 h in cells treated with a range of HgCl2 (Hg) or PbCl2(Pb) concentrations (0-10 μM). Exposure to Hg resulted in both time (F3,48=7.24; p=0.0004) and concentration- (F3,48=14.04; p<0.0001) dependent reductions in cell viability (Figure 5A). Dunnett’s test for posthoc comparison to a control (0 μM, 24 h) was used to determine the threshold for significant reductions. The highest concentration of Hg tested, 10 μM exhibited significant reductions in cell viability compared to control values (*p<0.05) at 48, 72 and 96 h. Exposure to Pb resulted in both time- (F3,47=19.89; p<0.0001) and concentration- (F3,47=4.29; p=0.003) dependent reductions in cell viability (Figure 5B). Pb (10 μM) exhibited significant reductions in cell viability compared to control values (*p<0.05) at 72 and 96 h, whereas the 1 μM group was significantly reduced compared to controls at 96 h. After examining the response to Hg and Pb, it appears that N2A cells were more sensitive to the concentration of Hg, whereas the length of exposure was a greater factor in Pb-induced reductions in viability.
Figure 5. Concentration- and Time-Dependent Changed in N2A Viability Following Exposure to Mercuric (Hg) and Lead (Pb) Chloride (0, 0.1, 1, or 10 μM). Exposure to Hg Resulted in Both Time- (F3,48=7.24; p=0.0004) and Concentration-(F3,48=14.04; p<0.0001) Dependent Reductions in Cell Viability (Figure 5A). Dunnett’s Test for Posthoc Comparison to a Control (0 μM, 24 h) was Used to Determine the Threshold for Significant Reductions.The Highest Concentration of Hg Tested, 10 μM Exhibited Significant Reductions in cell Viability Compared to Control Values (*p<0.05) at 48, 72 and 96h. Exposure to Pb Resulted in Both Time- (F3,47=19.89; p<0.0001) and Concentration- (F3,47=4.29; p=0.003) Dependent Reductions in Cell Viability (Figure 5B). Pb (10 μM) Exhibited Significant Reductions in Cell Viability Compared to Control Values (*p<0.05) at 72 and 96 h, whereas the 1 μM Group was Significantly Reduced Compared to Controls at 96 h. After Examining the Response to Hg and Pb, it Appears that N2A Cells were more Sensitive to the Concentration of Hg, whereas Length of Exposure was a Greater Factor in Pb-Induced Reductions in Viability. Data are Expressed as the Mean±SEM of N=4 Assayed in Duplicate.
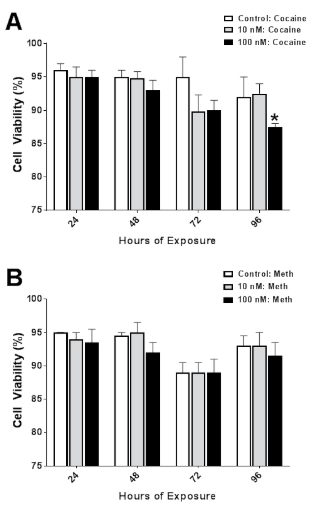
Cell Viability Following Psychostimulant: Time Course and Concentration Curve
To evaluate cytotoxicity, LDH activity was measured at four time points between 24-96 h in cells treated with a range of cocaine (COC) or methamphetamine (MA) concentrations (0-100 nM). Exposure to COC resulted in a time- (F3,36=4.62; p=0.008) dependent reductions in cell viability (Figure 6A). Dunnett’s test for posthoc comparison to a control (0 μM, 24 h) was used to determine the threshold for significant reductions. The highest concentration of COC tested, 100 μM exhibited significant reductions in cell viability compared to control values (*p<0.05) at only 96 h. Exposure to MA resulted in time- (F3,36=7.05; p=0.0008) dependent reductions in cell viability (Figure 6B). There were no differences in cell viability at any time point or concentration of MA compared to control values. Interestingly, few differences were observed following COC or MA exposure. Reductions in cell viability were dependent more on time of exposure than the concentration of COC or MA used. The observation of few concentration-dependent changes will facilitate the later studies when a time and concentration combination is used that will have minimal effects on cell viability. The rationale for this approach is to avoid any catastrophic damage to the cells, resulting in a more accurate representation of DAT expression and function.
Figure 6. Concentration- and Time-Dependent Changed in N2A Viability Following Exposure to Cocaine (COC) or Methamphetamine (MA) (0, 10, or 100 μM). Exposure to COC Resulted in Time- (F3,36=4.62; p=0.008) Dependent Reductions in Cell Viability (Figure 6A). Dunnett’s Test for Posthoc Comparison to a Control (0 μM, 24 h) was Used to Determine the Threshold for Significant Reductions. The Highest Concentration of COC Tested, 100 μM Exhibited Significant Reductions in Cell Viability Compared to Control Values (*p<0.05) at only 96 h. Exposure to MA Resulted in Time- (F3,36=7.05; p=0.0008) Dependent Reductions in Cell Viability (Figure 6B). There were No Differences in CellViability at Any Time Point or Concentration of MA Compared to Control Values. Data are Expressed as the Mean±SEM of N=4 Assayed in Duplicate
Determination of hDAT Density by [3H] GBR12935 Binding
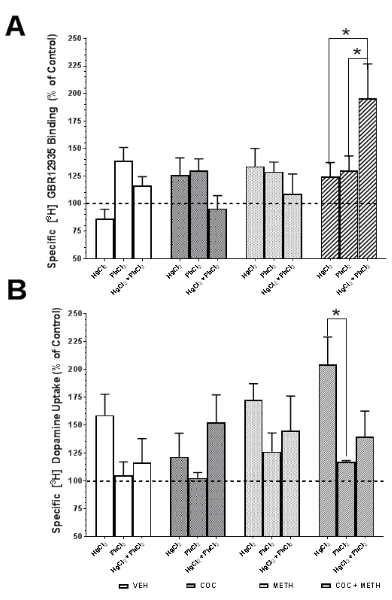
From the viability studies, the concentration of Hg and Pb was 10 μM and for COC and MA was 100 nM with an exposure time of 72 h. The density of hDAT was quantified by the binding of [3H] GBR 12935 to hDAT on the cell surface. Although metals, psychostimulants or combinations resulted in shifts in hDAT by ±30% compared to control values, many of these differences did not reach statistical significance (Figure 7A). There was a significant effect of stimulant on the hDAT density (F3,32=3.52; p=0.026) and a significant interaction between metal- and psychostimulant-mediated effects (F6,32=3.57; p=0.008). Exposure to treatment combinations resulted in elevated [3H] GBR12935 binding. Interestingly, density in the Hg+MA group increased by 161% over the control values. Also, the Hg+Pb+COC+MA treatment group mean increased 227% compared to control. Also, the Hg+Pb treatment group mean was 288% over the control mean values. These noted Hg-containing groups appeared to have an additive treatment effect on binding. Sidak’s test for posthoc multiple comparisons revealed significant increases in hDAT density in the COC-MA group comparing the Hg-Pb combination to just Hg or Pb alone (*p<0.05). We calculated the fractional occupancy of [3H] GBR12935 for DAT as well as the norepinephrine (NET) and serotonin (SERT) transporter using the formula: 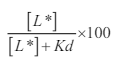
Where [L*] is the concentration of ligand used (50 nM) and the Kd (21 nM) is the reported affinity of the radioligand for the target. Using this formula, [3H] GBR12935 bound to a greater number of DAT > NET > SERT. Of the density of sites reported, 70% of the total number represents binding to the DAT, with only 18% and 0.8% of [3H] GBR12935 bound to NET and SERT, respectively. The use of GBR 12909 at a concentration of 5 μM displaced 99.99% of [3H] GBR12935 bound, leaving an accurate estimate of nonspecific binding. The reason this was done is to ensure that we were binding primarily to DAT and that we exceed the Kd value where only 50% of available DAT sites would be occupied.
Changes in hDAT Function Measured by Alterations in [3H] DA Uptake
From the viability studies, the concentration of Hg and Pb was 10 μM and for COC and MA was 100 nM with an exposure time of 72h. The functionality of hDAT was determined by measuring the uptake of [3H] dopamine into N2A cells via hDAT (Figure 7B). Similar to what we observed in the hDAT density assay, there was a moderate level of variability in the data. There was a significant effect of metal on dopamine uptake (F2,30=5.63; p=0.0084). The COC treatment group exhibited a 17% decrease in uptake compared to control values. The Hg+COC group also showed a 20% decrease. Interestingly, [3H] dopamine uptake studies showed the largest shift in treatment groups containing MA. Mean values in MA groups were increased 35-81% compared to control values. Sidak’s test for posthoc multiple comparisons revealed significant increases in hDAT density in the COC-MA group comparing the Hg alone to Pb alone with the COC-MA-Hg group being nearly 200% of control values (*p<0.05).
Relationship between hDAT Density and DA Uptake in N2A Cells Expressing hDAT
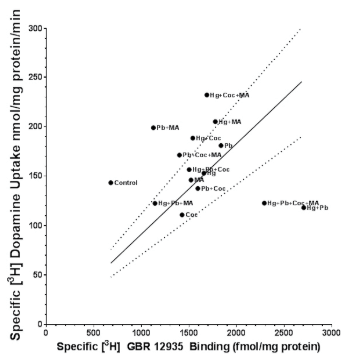
We examined the interaction/relationship between the hDAT density and function in N2A cells by plotting the data from the [3H] GBR12935 binding and [3H] dopamine uptake studies to determine if a relationship exists between the two variables (Figure 8). Surprisingly, the linear correlation between density and uptake was not significant. Instead, it appeared that exposure to either metals or psychostimulants, alone or in combination, resulted in a rightward shift of the curve. This shift represents an increasing density without the concomitant increase in uptake. Additional work is needed to determine if this change is an increase in density or possibly an increase in affinity of the hDAT for [3H] GBR12935.
Figure 8. Relationship Between hDAT Density ([3H] GBR 12935 Binding) and Uptake ([3H] Dopamine Uptake). The Basic Assumption is that Increasing Density of hDAT shouldLead to an Increase in DA Uptake. Deviations from this Relationship would Suggest Changes in Protein Processing or Function. Observations Compared to Control Values Reveal a Rightward Shift in the Data (Higher Density with Minimal Changes in Uptake) whereas a Few Groups (Predominantly the Hg Groups) are Shifted to the Right as well as Upward. This Could Suggest the Increase in hDAT Density Results in Elevated DA Uptake Due to the Increased Density of a Function hDAT Protein on the Cell Surface. Data is Taken From the Binding and Uptake Data Presented in Figure 7
DISCUSSION
The DAT is the principal target for widely abused psychostimulants such as COC and MA. The reinforcing properties of these drugs (which likely underlie their addictive properties) are strongly correlated with their affinities for the DAT.49 DA release in the nucleus accumbens is believed to be the main mediator of the reinforcing and locomotor activating properties of psychostimulants.50 COC binds to the Na+ binding site on the DAT and alters the Cl– binding site, thus preventing the binding of both ions. Because translocation of DA across the membrane of the pre-synaptic neuron is inhibited, there is an increased extracellular DA concentration. Under normal physiological conditions, the DAT transports extracellular DA back into the nerve terminal (i.e., uptake). However, within minutes after MA administration, non-vesicular DA efflux occurs through reversal of the DAT creating high extracellular DA concentrations. Of interest, blockade of DAT by COC leads to a rapid increase in DA uptake in synaptosomes prepared from treated rats, a preparation from which the drug has presumably been washed out.35 Perhaps this occurs via enhanced recruitment of DATs to the plasma membrane.12 These increases in DA uptake and cell surface expression, observed in rodents and cell lines, respectively, after acute COC administration likely represent efforts to maintain normal synaptic DA functions. In humans who have repeatedly increased synaptic DA levels through the use of COC, increased DAT function is also observed, as assessed in synaptosomes from cryoprotected human brain.11 It is possible that an overabundance of extracellular DA triggers this compensatory increase in DAT activity, which would ultimately produce a deficit in extracellular DA, perhaps contributing to drug dependence. From the in vivo and in vitro studies comes our interest in whether environmental factors (heavy metal exposure) and psychostimulant (ab) use could exhibit overlapping mechanisms leading to increased addictive potential or increased toxicity of either factor.
There is evidence that metals may inhibit or increase the binding of ligands to receptors in vitro.37,38,43,45,46,51,52,53 The objective of this study was to investigate heavy metal exposure effects on psychostimulant binding at the DAT. This is an especially pertinent issue given that Hg and Pb uniquely threaten sub-populations where drug abuse is more common, such as urban minorities, or individuals of lower socio-economic status. Experiential elements, availability, drug history, poverty, etc., must be considered in the list of dispositional factors that determine drug habits in humans. However, it also must be considered that other types of environmental events may contribute to the abuse potential of selective drugs. That is, to the extent that Hg, Pb, or any other xenobiotic chemical alters the impact of a set delivery of a drug, motivational features related to drug seeking and taking may be redefined and influence maintenance drug use or the effectiveness of certain pharmacotherapies for drug abuse.
We have been able to express hDAT in both a transient and stable manner. In N2A cells, we have generated a cell line of neuronal origin that stably express hDAT and allow for studies into the regulation of hDAT production, processing, and function. Tagging hDAT with GFP allows for visualization of changes via microscopy and anatomical references regarding the location of hDAT within the cell. Our N2A cell line expressing hDAT has been invaluable in examining cellular changes in hDAT regulation. The molecular mechanisms associated with metal-induced neurotoxicity is complex and metal-dependent. For each metal, there are sites or processes, which are the most vulnerable and thus, primarily affected. Hg exerts its toxic effect by binding to the thiol groups on proteins, including the DAT.54 The DAT has a disulfide bridge that is important for the conformational structure of the transporter.54 After Hg binds to this bridge, there is a conformational change in DAT affecting its normal functioning. Cells adapt by processing more DAT and increase the trafficking rate to get DAT to the cell surface.55 Our findings support these reports of increased DAT density and function.13 In the presence of metal ions such as Hg2+, binding to DAT does not follow a classic monotonic response. At lower concentrations, density is elevated, but at higher concentrations of Hg2+ binding of [3H] WIN 35,428 is reduced.13,47 It is possible that our concentrations of Hg were too low since we chose a concentration based on its lack of toxicity after 72 hours of exposure. We observed a general increase of approximately 20-25% in the density of hDAT as measured by [3H] GBR12935 binding. This coincided with a 25-50% increase in [3H] DA uptake. Pb competes with calcium-mediated synaptic vesicle release resulting in an increased DA release from the synapse.56 In general, investigators have reported that Pb exposure results in DA hypofunction.7,8,9 In vivo studies have demonstrated that Pb exposure can result in a reduction in binding to DAT8 and a decrease in DA uptake.9 Our findings suggest that neuronal cells strive to maintain DA clearance by increasing the density of DAT at the plasma membrane. Reasons for the discrepancy between in vivo and in vitro studies include; the chemical form of Pb, the dose or concentration of Pb used, the time of exposure, and differing model systems. Most in vivo studies use Pb-acetate due to improved solubility, but this organic form of Pb exhibits greater toxicity compared to the inorganic form, Pb-chloride, used in the present studies. Another potential source of variation between studies is the dose or concentration used. We used a concentration of 10 μM Pb-chloride which equates to approximately 2.8 ppm. The study by Noureddine et al9 utilized a dose of 1000 ppm, nearly 400-fold higher than our studies. Behavioral effects of Pb appear with blood Pb levels exceeding 10 μg/dL,7 which correlates with a concentration of 0.1 ppm. Pb responses may follow the changes in DAT density observed following Hg exposure. Responses that appear ‘biphasic’ or non-monotonic, it is possible that lower concentrations of Pb increase the density of DAT and uptake of DA, and higher concentrations (with longer exposure times) result in DA hypofunction. Thus, exposure to sub-toxic concentrations of heavy metals results in an indirect up-regulation in DAT density by promoting an increase in extracellular DA. When examining the magnitude of change comparing Hg to Pb, changes in hDAT density were similar. When investigating changes in uptake, a clear difference in the response magnitude of Hg-mediated changes in DA uptake was observed compared to the Pb group. The combination of Hg+Pb was slightly higher than Pb alone. Only in the COC+MA group did these differences in density and uptake reach statistical significance.
Under normal conditions, the majority of DAT protein is found at the cell surface. The translocation of DAT from the membrane to the cytosolic space is a fundamental mechanism in the regulation of DAT homeostasis and functioning. Cell cultures expressing cloned DAT demonstrate that treatment with psychostimulants regulates DAT expression in the plasma membrane.36,55,57,58 Psychostimulants may act by promoting exocytosis of internalized DAT and/or by decreasing constitutive internalization of DAT and thus increasing plasma membrane DAT by altering the balance of internalization and recycling to the surface. Cell surface redistribution of DAT is a mechanism that contributes to the enhancement of extracellular DA levels in response to psychostimulants. The COC treatment group and the Hg+COC treatment group means showed a trend towards decreased uptake compared to control. Yet, binding was elevated in COC treated cells. Because COC is a DAT inhibitor, [3H] DA uptake is blocked leading to increased synaptic DA, and increased postsynaptic stimulation. With the dopaminergic system in a compromised state during COC exposure, the up-regulation of the DAT could reflect a homeostatic response whereby increased capacity for DA reuptake would maintain neurotransmission at more normal levels. Thus, these data suggest that the DAT might actively participate in modulating the behavioral consequences of chronic COC exposure. There are conflicting reports, however, concerning changes in DAT function as a mechanism contributing to addiction. Some studies have found changes consistent with the reduced activity of DAT, including reduced DA uptake,28 down-regulation of DAT59 and elevated extracellular fluid (ECF) concentrations of DA.30 In contrast, other groups have reported changes consistent with increased activity of DAT, including increased DA uptake35,60,61 and an attenuation of COC-induced increases in ECF levels of DA.62 The variable effect of COC on uptake likely reflects the use of different dosing regimens, routes of administration, brain regions, ECF concentration of DA at the time of COC administration, as well as the techniques to quantify DAT function.2,32,60 [3H]DA uptake studies showed the largest shift in treatment groups containing MA. MA exposure elicited a marked up-regulation in both DAT binding and uptake. This is what is expected since MA promotes DA release.15 The presence of MA may initiate a compensatory mechanism whereby DAT density is increased to enhance removal of DA from the synaptic cleft.
Figure 7. Effects of Metals and Psychostimulants Alone or in Combination on hDAT in N2A cells. From the Viability Studies, the Concentration of Hg and Pb was 10 μM and for COC and MA was 100 nM with an Exposure Time of 72 h. The Density of hDAT was Quantified by the Binding of [3H] GBR 12935 to hDAT on the Cell Surface. Although Metals, Psychostimulants or Combinations Resulted in Shifts in hDAT by ±30% Compared to Control Values, Many of These Differences did not reach statistical significance (Figure 7A). There was a significant effect of stimulant on the hDAT Density (F3,32=3.52; p=0.026) and a Significant Interaction between Metal- and PsychostimulantMediated Effects (F6,32=3.57; p=0.008). Sidak’s Test for Posthoc Multiple Comparisons Revealed Significant Increases in hDAT Density in the COC-MA Group Comparing the Hg-Pb Combination to just Hg or Pb Alone (*p<0.05). Functionality of hDAT was Determined by Measuring the Uptake of [3H] Dopamine into N2A Cells via hDAT (Figure 7B). Similar to what we Observed in the hDAT Density Assay, there was a Moderate Level of Variability in the Data. There was a Significant Effect of Metal on Dopamine Uptake (F2,30=5.63; p=0.0084). Sidak’s Test for Posthoc Multiple Comparisons Revealed Significant Increases in hDAT Density in the COC-MA Group Comparing the Hg Alone to Pb Alone with the COC-MA-Hg Group being Nearly 200% of Control Values (*p<0.05). Data are Expressed as the Mean±SEM of N=4 Assayed in Duplicate.
Our study demonstrates that metal/drug treatments increase the cell surface distribution of DAT and that this redistribution of DAT could be associated with increases in extracellular DA. This relationship was shown in Figure 7A with a significant interaction between metal and psychostimulant (F6,32=3.57; p=0.008). Because changes in DA by psychostimulants, drug-related trafficking of DAT may be an important mechanism in the development of its abuse. Increased GBR binding with no difference or decreased DA uptake may suggest decreased function of DAT. In turn, this will result in decreased DA uptake leading to increased synaptic DA. If not cleared properly, high levels of synaptic DA may lead to cell damage over time and further changes in DAT density and function. Collectively, heavy metals such as Hg and Pb have been shown to be directly toxic via multiple pathways like oxidative stress, or protein interactions can also exert indirect toxic effects. The data reported in this study would suggest an indirect role of metal toxicity regarding the effects on DAT function and synaptic DA levels.
CONCLUSION
The impact of our findings accounts for potential environmental factors, such as heavy metal exposures, that could facilitate the abuse of psychostimulants, or increase toxicity. We found that Hg, Pb, COC and MA all exert some effect on hDAT through changes in density or uptake of DA. Interestingly, the effects do not follow a simple additive, synergistic, or antagonistic mechanism. We would have expected additive or synergistic responses in most instances. The fact that this is not observed suggests a more complex, which is both direct and indirect; pathway is involved in heavy metal or psychostimulant alteration in DAT function. A generalization of the results does show an increase in both density and uptake, which is a response to increased DA in the synapse. There were no explicit relationships between individual exposures to Hg, Pb, COC or MA, and no significant relationships when transfected N2A cells were exposed to combinations of heavy metals and psychostimulants. From the forensics perspective, lower doses of a psychostimulant could lead to an exaggerated response due to the pre-exposure and DA modifications associated with heavy metals. We will target future in vitro studies at determining DAT turnover and the treatment effect on N-linked glycosylation of DAT. Additional molecular studies are underway investigating epigenetic changes following extended exposure to Hg and Pb. If genetic changes occur, it would have a lasting impact on the responsiveness to psychostimulants. In vivo studies involve administering metals in drinking water for several weeks and measuring DAT changes months later. Chronic exposure to psychostimulants has been shown to elicit long-term neurochemical changes in neurotransmitter systems manifesting as tolerance or epigenetic changes. The restriction on in vitro studies is only ‘acute’ exposure can be examined.
ACKNOWLEDGEMENTS
The authors would like to acknowledge the expertise and assistance of Dr. Greg Sawyer and Crystal Shults who aided in the construction of the transfected N2A cell lines. The work supported in part by subcontract flow-thru funds from NIH DA13137 and through intramural funding by the Oklahoma State University Center for Health Sciences.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.


