INTRODUCTION
The existence of the adrenal hormone cortisol was first postulated by Phillip Hench and Edward Kendall in 1930. Using a combination of clinical conjectures and 3,000 lbs. of animal adrenal gland tissue,1 they believed there existed a substance that was produced in biological species in response to physiological stress. By 1940 Kendall had isolated cortisol and dubbed it “substance E”, as had Tadeus Reichstein-Kendall’s competitor working simultaneously in Switzerland. The two joined forces thanks to Merck & Company as rumor had it that the Nazi’s were secretly shipping tons of bovine adrenal tissue from Argentina via submarines and looking to militarize adrenal hormones to optimize their pilots and soldiers.1 In 1941 The United States government and National Research Council prioritized the production of stress hormones ahead of penicillin development and antimalarial research.1 By 1948 Merck Pharmaceutical Company had invested $13 million dollars ($130 million adjusted for inflation in 2016) in “substance E” and had neither military nor clinical applications until September 1948. Hench and Kendall requested Merck to give them 5 grams of substance E to salvage their project. Hench and Kendall decided to give 50 mg intramuscular injection of “substance E” to a patient suffering from rheumatoid arthritis at Mayo Clinic in Rochester, Minnesota known as Mrs. G. Initially bedridden due to pain, by the seventh day of treatment Mrs. G was shopping in downtown Rochester claiming, “I have never felt better in my life”.1 In October of 1950 Hench, Kendall, and Reichstein won the noble peace prize in medicine for “investigations of the hormones of the adrenal cortex”. However, Mrs. G became increasingly Cushingoid as time went on and it became clear that cortisol was not the panacea that it had promised.1 Since then, glucocorticoid therapy has become a staple drug for physicians and has helped save countless lives. Modern medicine has taught us the importance of tapering off steroids and the significant adverse effects associated with long-term use of glucocorticoids. Glucocorticoids have gained unprecedented importance in treating autoimmune and inflammatory disease, with one study conducted between 1998-2008 suggesting that 1.2% of Americans over the age of 20 were at one point on glucocorticoid therapy, accounting for approximately 2.5 million Americans over the course of a decade.2Given the immunomodulatory clinical value and ubiquitous presence of glucocorticoids, this paper seeks to explore the mechanisms by which glucocorticoid therapy induced alterations to the hypothalamic-pituitary-axis (HPA) could cause secondary hypogonadism.
Hypogonadotropic Hypogonadism
Patients presenting with hypogonadism may be a more prevalent phenomenon in the general medical care setting than most clinicians readily realize. Hypogonadism most commonly presents as a comorbidity in obese patients and those with type 2 diabetes mellitus. The etiology of hypogonadism can be divided into congenital (primary) or acquired (secondary). Male patients suffering from hypogonadism will experience symptoms of low libido, fatigue, erectile dysfunction, decreased testosterone, reduced facial hair growth, loss of muscle mass, decreased the testicular size, and gynecomastia. Female patients will present lack of menstruation, slow or absent breast growth, hot flashes, loss of body hair, reduced or absent sexual drive, or milky discharge from breasts.3 Adult-onset hypogonadotropic hypogonadism is characterized in women by secondary amenorrhea, decreased libido, infertility, and osteoporosis.3 However, measuring estradiol levels in females varies with regard to where the patient is in their respective menstrual cycles.3 With respect to exogenous glucocorticoid use, we are concerned with acquired, or secondary, or also known as hypogonadotropic hypogonadism (HH). It is well-documented that severe illness, stress, malnutrition, and excessive exercise can cause reversible suppression of gonadotropin release. In men, true hypogonadism may be distinguished from age-related decline and erectile dysfunction via laboratory evaluation of morning total testosterone. Specifically, free testosterone (testosterone that is not bound to sex hormone-binding globulin, SHBG) should be measured as this testosterone is bioavailable. Morning measurements of testosterone <300 ng/dL in combination with symptoms described above suggest a testosterone deficiency. Very low morning testosterone levels below 150 ng/dL qualify patients for pituitary magnetic resonance imaging (MRI). One cross-sectional study of 2,162 men aged 45 and older at primary care facilities across the United States found that 38.7% of them had serum testosterone levels below 300 ng/dL.4 If testosterone deficiency has been determined, luteinizing hormone (LH) and follicle stimulating hormone (FSH) may be measured to elucidate a primary or secondary cause in origin. High-levels of LH and FSH suggest a primary cause (i.e. Klinefelter, HIV, cryptorchidism, chemotherapy, radiation, etc.) and low LH and FSH levels indicate a secondary cause at the level of the hypothalamic-pituitary axis. Evidence suggests that chronic long-term glucocorticoid therapy causes deviation in the HPA axis and results in secondary, hypogonadotropic hypogonadism (Figure 1 ).3-5
Figure 1. Testosterone Suppression by Glucocorticoids
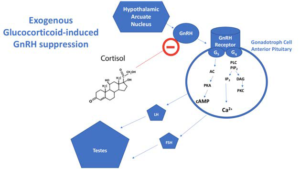
Gonadotropin Releasing Hormone (GnRH) Axis
GnRH neurons arise from embryonic neural crests cells of the ectoderm layer of the embryo and migrate from the olfactory placode to the hypothalamus. During puberty, sufficient adipocyte reserves signal increases in Leptin, which leads to a pulsatile increase of the neuropeptide kisspeptin. Puberty marks the initiation of a pulsatile GnRH release and subsequent mature adult functioning of the gonads. This recently discovered Kisspeptinneurokinin-B-dynorphin pathway acts as an upstream regulator of the GnRH-LH/FSH pathway. It is in the hypothalamus that the GnRH neurons release their GnRH vesicles into the hypophyseal portal capillary bloodstream which carries the GnRH down to the gonadotropic cells of the anterior pituitary. Once gonadotropic cells come into contact with GnRH, they release LH and FSH into the circulatory system. LH and FSH have trophic effects on the male testes and regulate the female menstrual cycle. The adipocytekisspeptin-GnRH-LH-FSH axis can be manipulated and altered in certain populations of patients receiving long-term glucocorticoids. We look to explore all the possible biochemical mechanisms by which exogenous glucocorticoids could potentially alter the HPA axis and induce hypogonadotropic hypogonadism (Figures 1 and 2).3-5
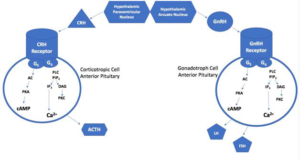
Figure 2. Corticotropin and Gonadotropin Production in HPA
Glucocorticoids and Hypothalamic-Pituitary Suppression
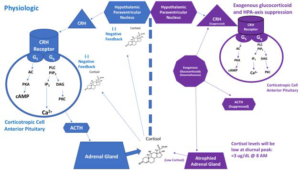
The highest prevalence of glucocorticoid receptors is found in the hypothalamus and hippocampus of the human brain.5 It is possible that chronic exogenous glucocorticoids could have multiple receptor interactions in the hypothalamus, particularly on GnRH neurons. Previous studies have shown that states of elevated glucocorticoid, regardless of whether it is endogenous or exogenous in origin can cause hypogonadism in both mice and humans.6 This study showed that exogenous dexamethasone suppresses gonadotropins in both castrated and non-castrated mice. Castrated mice would not have testosterone and therefore no negative feedback, and therefore castration serves as a powerful stimulus for elevating levels of GnRH, GnRH receptor, LH, and FSH. However, pharmacological doses of dexamethasone were shown to suppress post-castration increases in GnRH-R, GnRH, LH, and FSH levels.6 Another study conducted in 2010 on mice demonstrated overlapping biochemical processes between glucocorticoid-induced and hypogonadotropic-induced myopathy in mice.7 Gene expression analysis revealed the dephosphorylation of the FoxO gene family by dexamethasone injection.7 The study also showed dexamethasone-induced animal model of muscle atrophy and signaling effects in the PI3K/Akt pathway which led to myopathy but also happened to show significantly reduced size of the prostate and seminal vesicles in the mice (Figure 3).7
Figure 3. Suppression of HPA by Glucocorticoids
Glucocorticoids Effect on Mitochondria
Although glucocorticoids effect on mitochondria can be neuroprotective at low doses, chronic high-dose glucocorticoid administration was shown to alter mitochondrial oxidation and calciumholding capacity and to produce neuronal inhibition as well as an allostatic overload of neurons.8 It could be that over time glucocorticoid receptor activation on GnRH neurons induces apoptosis and loss of GnRH pulsatility. It is also important to note that androgen and steroid hormone synthesis occurs inside the mitochondria. Dehydroepiandrosterone (DHEA), the precursor to testosterone and estrogen is synthesized by a battery of enzymes specifically found inside the mitochondria of the zona reticularis of the adrenal cortex. These studies hint at possible insults and injuries to GnRH neurons and how chronic exogenous administration of glucocorticoids could be hypogonadotropic to patients via their deleterious effects on mitochondria.
Glucocorticoids and Adipogenesis
Glucocorticoids have a potent stimulatory effect on terminal adipogenesis as seen in patients with Cushing “buffalo hump” cervical fat pad.2 Leptin is produced in direct proportion to adipose tissue. This glucocorticoid-induced increase in adipose tissue can cause increased leptin and alter the sensitive pulsatility of kisspeptin production causing subsequent dysregulation of the GnRH-sex hormone axis.
Endogenous Glucocorticoidsand Circadian Variations
Aside from the tangible distributions of glucocorticoid receptors and GnRH neurons, it may be consequential for the clinician to take into consideration intangible factors such as time, pulsatility, and natural circadian variations in endogenous glucocorticoids9 when prescribing long-term glucocorticoid therapy. It may be that exogenous long-term glucocorticoid therapy has unwanted alterations in GnRH neurons pulsatility and subsequent hypogonadotropism may be inadvertently induced. In a similar fashion, Leuprolide given in a continuous manner disrupts the pulsatility of the GnRH neurons.
CRH-ACTH-Cortisol Axis
Cortisol is an endogenous, 21-carbon, four-fused ring, a hydrophobic steroid that is produced in zona fasciculata- the middle layer of the adrenal cortex which comes from mesodermal embryologic origins. In response to stress, corticotropin-releasing hormone (CRH), a 41 amino acid neuropeptide is released from CRH neurons in the hypothalamus. The highest density of CRH neurons are found in the parvocellular neurons of the hypothalamic paraventricular nucleus (PVN), but CRH mRNA can also be found outside the hypothalamus in bed nucleus of the stria terminalis, the central nucleus of the amygdala, locus coeruleus, cerebral cortex, cerebellum, and dorsal root neurons of the spinal cord as well as chromaffin cells of the adrenal medulla, sympathetic ganglia of the autonomic nervous system, and non-neural peripheral organs such as immune cells, skin, and gastrointestinal tract.10 It is interesting to note that approximately 50% of parvocellular CRH neurons in the PVN of the hypothalamus also release a small amount of vasopressin as well as CRH in response to stress. From here, the CRH travels down the hypophyseal capillaries to the anterior pituitary gland where it binds its receptor on corticotropin cells. There are two types of CRH receptor, 1 and 2, both of which are Gproteins coupled to adenylate cyclase which subsequently increase intracellular levels of cAMP (Figure 2).10 This increase in intracellular cAMP in corticotropin cells of the anterior pituitary gland causes transcription of proopiomelanocortin (POMC) mRNA which encodes a 241 amino acid precursor protein POMC. POMC is cleaved into α-melanocyte stimulating hormone (α-MSH), adrenocorticotropic hormone (ACTH), and β-endorphin. Of these three neuropeptides, ACTH travels through the systemic circulation to its receptors, the melanocortin type 2 receptor (MC2R), in the zona glomerulosa and the zona fasciculata. The ACTH receptor, MC2R is also a cell-membrane associated G-protein linked to adenylate cyclase, which also increases cAMP and activates protein kinase A (PKA).11 ACTH induced an increase in cAMP leads to an upregulation of the genes encoding for mitochondrial cytochrome oxidase p-450 enzymes, specifically steroid 11B-hydroxylase.11 This enzyme, abbreviated as CYP11B1, localizes to the inner mitochondrial membrane and converts 11-deoxycortisol to cortisol in both the zona glomerulosa and the zona fasciculata, as well as converting 11-deoxycorticosterone to corticosterone in the zona glomerulosa.11 Although this enzyme is present in both the zona glomerulosa and the zona fasciculata, the synthesis and release of cortisol are more prominent in the zona fasciculata while aldosterone synthesis takes priority in the zona glomerulosa. Approximately 80% of cortisol is bound to cortisol-binding globulin (CBG) in circulation, with the rest of it being free or weakly bound to albumin. Apart from fish, all other vertebrate classes have a plasma protein that binds glucocorticoids with high affinity, and the primary structure of CBG defines it as a clade A serine proteinase inhibitor (SERPINA).12 A 2014 study has shown that CBG is the primary determinant of circulating plasma cortisol levels in humans.13 From here cortisol can go on to have its systemic effects such as increased blood glucose through gluconeogenesis, suppressing the immune system, aiding in the catabolism of muscle, as well as inhibiting fibroblasts and bone formation.14-15
DISCUSSION
In summary, evidence suggests that chronic long-term glucocorticoid therapy causes deviation in the HPA axis and results in secondary hypogonadotropic hypogonadism (HH) in males and females (Figures 1 and 3). In this review (limited to discussion of hypogonadism in males), we have explored the possible biochemical mechanisms by which exogenous glucocorticoids could potentially alter HPA axis and induce HH. Aside from the tangible distributions of glucocorticoid receptors and GnRH neurons, it may be consequential for the clinician to also take into consideration intangible factors such as time, pulsatility, and natural circadian variations in endogenous glucocorticoids9 when prescribing long-term glucocorticoid therapy. It may be that exogenous longterm glucocorticoid therapy has unwanted alterations in GnRH neurons pulsatility and subsequent hypogonadotropism may be inadvertently induced.
The prevalence of low testosterone concentrations is high in a number of clinical conditions, especially in chronic diseases and the effect would be more prominent when treatment requires medications that exacerbate this condition, namely glucocorticoids. If the clinical manifestations consistent with androgen deficiency are present, testosterone measurement should be performed. Because a substantial amount (30-40%) of circulating testosterone is bound tightly to sex hormone binding globulin (SHBG), glucocorticoids cause suppression of SHBG and lower the total testosterone sometimes too low normal levels, thus measurement of free and bioavailable testosterone is mandatory to elucidate the cause.16
Men with rheumatic diseases, notably the systemic rheumatic disorder rheumatoid arthritis, may manifest the clinical symptoms and laboratory manifestations of hypogonadism in up to 30% of cases.17 Hypogonadism in these patients may be due in part to systemic inflammation with elevated cytokines (such as interleukin-1 (IL-1) and tumor necrosis factor alpha (TNF-α)), complications of rheumatoid lung, and finally treatment with glucocorticoids. Studies showed that hypogonadism may occur early in the course of disease in the absence of complications and even glucocorticoid therapy.18 In men with long-standing rheumatoid arthritis, low free testosterone fails to normalize after the suppression of inflammation with anti-TNF therapy,18 on the other hand, while testosterone therapy improves symptoms of hypogonadism it does not reduce inflammatory arthritis disease activity.19
Men with systemic lupus erythematosus may demonstrate low free testosterone level consistent with both primary and secondary hypogonadism. Gonadotropin suppression and hypogonadism may be induced by systemic illness and inflammation, major organ involvement or failure (such as brain, heart, lung, and kidney), and/or glucocorticoid therapy.19-20
CONCLUSION
In conclusion, while management of functional causes of hypogonadism might improve clinical and biochemical hypogonadism, in many instances, however, where functional causes of hypogonadism cannot be treated and patients require long-term glucocorticoid therapy for chronic comorbid illnesses, testosterone therapy can be considered in order to improve quality of life and prevent secondary complications of androgen deficiency.
Given the nature of autoimmune/rheumatologic diseases and the proclivity towards requiring long-term glucocorticoids for treatment, it would be important for physicians, especially rheumatologists, to be knowledgeable of the wide spectrum of intended and unintended adverse effects of glucocorticoid therapy. Understanding the biochemical pathways underlying the unintended adverse effects of exogenous glucocorticoids in causing hypogonadotropic hypogonadism may allow for better patient education prior to starting treatment and prevent unexpected surprises for patients (on this potentially sensitive to discuss the issue, especially for men) undergoing long-term glucocorticoid therapy. A greater understanding of these biochemical pathways/ mechanisms may also lead to potential new counter-therapies by pharmaceutical companies to engineer new medications or treatments that may prevent these unwanted consequences of longterm glucocorticoid therapy.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.


