INTRODUCTION
GD is an inherited autosomal recessive metabolic defect due to a deficiency in the lysosomal enzyme b-glucocerebrosidase, which leads to deposition of glucocerebroside in cells of the macrophagemonocyte system, predominantly in the spleen, liver, and bone marrow. There are three clinical types, including type 1 (GD1), the non-neuronopathic type; type 2 (GD2), the infantile-onset, an acute neuronopathic type and type 3 (GD3), the juvenile-onset, a chronic neuronopathic type 1.1 The third type was divided into three subgroups (GD3a, GD3b and GD3c) on various clinical features. In GD3a, patients exhibit as progressive myoclonic epilepsy, with or without horizontal supranuclear gaze palsy, and mild systemic findings. A young man of GD3a we reported as follows, who was treated only as myoclonic seizures at first attending our neurology clinic, while he had been diagnosed as GD years ago.
CASE REPORT
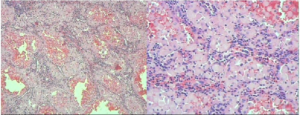
An 18 year-old boy with main complaint of convulsions in the left upper limb came to see a neurology doctor in our hospital. The left upper limb tic occurred several to dozens of times a day, and each time just lasted for seconds. And he was diagnosed as myoclonic seizures, with lamotrigine 25 mg once a day and a Electroencephalographic (EEG) examination. The routine EEG revealed generalized nonrhythmic paroxysmal rapid spikes with occipital predominance increased by photic stimulation and normal background activity. The frequency of spikes increased in harmony with the frequency of flicker (up to 25 Hz) and spikes frequently occurred on eye closure (Figures 1A, 1B and 1C). The EEG neurologist got a medical history that his convulsions developed after the splenectomy about 3 years ago, and he had been diagnosed as GD before. He was first discovered to have an enlarged spleen because of a cold and was diagosed with “GD combined hepatosplenomegaly” (without details) in Guangdong Provincial People’s Hospital since 6 years old. He was suggested to take enzyme replacement therapy (ERT), but not accepted it for lack of money. In the decades that followed, he took traditional Chinese medicine, and rechecked by B-ultrasonic irregularly still with hepatosplenomegaly. His β glucosidase levels was considerably subnormal (without accurate number recorded), which can be diagnosed with GD, tested in Peking Union Medical College Hospital on December 17th, 2010. He was a Chinese and born with a parents with no consanguineous marriage. He was delivered after full-term normal pregnancy. Development of the child was normal. There was no history of easy bruising or prolonged bleeding on trauma, hematemesis, fever, night sweats, and weight loss or bone pains. His father’ brother had splenomegaly with unknown reasons. His other family members, including his parents and two siblings, were normal. 3 years ago, he was admitted into our hospital, the chief complaint was splenomegaly for 10 years and left abdominal pain for 2 days. On physical examination, he looked malnourished; however there was no icterus or lymphadenopathy. He had firm, non tender massive splenomegaly and no hepatomegaly. There were no signs of ocular motor problems or other neurological abnormalities. The patient and his family denied any apparent intellectual decline noticeable in his daily life. Rest of systemic examination was essentially normal. Lab investigations revealed bicytopenia (hemoglobin=9.9 g/dl, white blood cells=3.17*109/L and platelets=73*109 /L). Liver enzymes were normal (aspartate aminotransferase=34I U/ ml, alanine aminotransferase=19 IU/ml), and the same as serum proteins and albumin, kidney function test and urine analysis. PT (Prothrombin Time) was 15.3 s [International Normalized Ratio (INR)=1.29] and Partial Thromboplastin Time (PTT) was 50.5 s. Hepatitis B Virus (HBV), Syphilis and HIV (human immunodeficiency virus) antibody test were negative. Computed Tomography (CT) revealed grossly enlarged spleen (247*140 mm) [Figures 2A, 2B] and splenic and portal veins had large diameter. Bone marrow [Figure 3] aspiration was performed which revealed Gaucher cells in a background of proliferous erythroid, myeloid and megakaryocytic lineage cells. Splenectomy was performed and one section completely capsulated, soft and dark red, partially drab yellow, were sent to pathological biopsy. Lots of Gaucher cells were seen in spleen sections (Figure 4). Final diagnosis was GD3a.
Figure 1: Generalized nonrhythmic paroxysmal rapid spikes and normal background activity are observed in the routine EEG examination of the patient (1A).
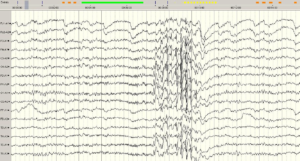
There is marked eye closure sensitivity (1B).
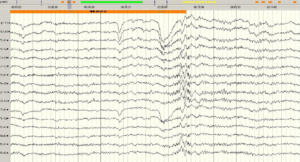
The frequency of the flicker is 9-22Hz (1C).
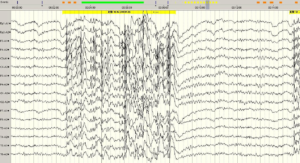
Figure 2: A. US and B. CT show grossly enlarged spleen.
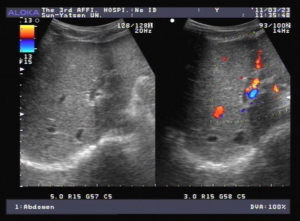
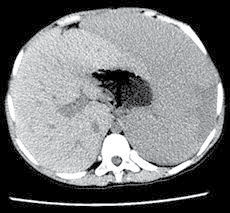
Figure 3: Gaucher cells in marrow smear (Wright Giemsa stain*100).
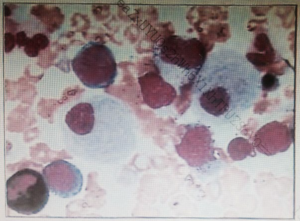
Figure 4: Lots of foamy cells were seen in spleen sections.
DISCUSSION
GD is inherited as autosomal recessive traits with a overall incidence of approximately 1 in 100,000 live births.1 GD is characterized by accumulation of Glucosylceramide (GC) in the cells of monocyte/macrophage system located in liver, spleen or central nervous system called Gaucher cells, which causes splenomegaly or epilepsy. And the glucocerebrosidase gene located on chromosome 1q21 and consists of 10 introns and 11 exons2 has been described in the GBA gene region with more than 300 mutations, including point mutations, deletions, insertions, splicing aberrations and various rearrangements.
Gaucher disease can also present as hydrops in the perinatal period which is often lethal. There may be congenital ichthyosis, also known as collodion baby.3 GD should therefore be considered as a differential diagnosis of any hydrops in pregnancy. Diagnosis of GD is made on the basis of clinical history, physical examination, laboratory test and confirmed by a blood test showing deficient glucocerebrosidase enzyme and genetic mutation studies when the diagnosis is doubtful.
The case we reported onseted with splenomegaly at the age of 6 and continued to enlarge in the last years. Thrombocytopenia, anemia and leucopenia are seen on the blood counts. Liver enzymes were normal and PT and PTT were prolonged a little without an increased tendency to bleeding. His β glucosidase levels was considerably subnormal (without accurate number), that measurement of glucocerebrosidase enzyme activity in leucocytes or skin fibroblasts on a skin biopsy is the gold standard for diagnosing Gaucher disease.4 The patient’s biospies of bone marrow and spleen all supported diagnosis of GD. EEG findings in our case demonstrate a similar pattern, as a case report before,5 with rapid spike activity, photosensitivity in harmony with the flicker frequency and eye closure sensitivity, which is few reported in GD.
ERT is the standard of care for patients of GD 1 and GD 3.6 ERT is most effective in reducing the liver and spleen size and the bone symptoms, and improving blood counts. When there is no access to expensive ERT, bone marrow transplants and liver transplants may be an option, albeit inferior to ERT. Splenectomy is rarely indicated for palliation for more than 15 years in Western with a higher risk of cholesterol gall stones, pulmonary complications including pulmonary hypertension, avascular necrosis of bone and iron overload states.7,8,9,10 The patient here didn’t have enough money to receive ERT at the beginning and had splenectomy 3 years ago to relieve pain. Then the disease progressed and developed myoclonic epilepsy. It can be expected that, without ERT, the patient’s symptoms will be worse and with new symptoms.
CONCLUSION
GD is so rare that it is easily missed diagnosis or misdiagnosis in the clinic. Patients presented with myoclonic seizures should be considered in the differential diagnosis of those with unexplained splenomegaly especially with an extended period of time. It is importance to collect his detailed medical and family history, and have him done some essential laboratory tests and auxiliary examinations.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
CONSENT
The patient has provided written permission for publication of the case details.


