1. World Health Organization (WHO). Diabetes. Global report on diabetes. Website. http://www.who.int/diabetes/global-report/
2. World Health Organization (WHO). Obesity and overweight. Website. http://www.who.int/mediacentre/factsheets/fs311/en/
3. Das AK, Shah S. History of diabetes: from ants to analogs. J Assoc Phys India. 2011; 59 Suppl: 6-7.
4. Templeman NM, Skovso S, Page MM, Lim GE, Johnson JD. A causal role for hyperinsulinemia in obesity. J Endocrinol. 2017; 232(3): R173-R183. doi: 10.1530/JOE-16-0449
5. Mehran AE, Templeman NM, Brigidi GS, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012; 16(6): 723-733. doi: 10.1016/j.cmet.2012.10.019
6. D’souza AM, Johnson JD, Clee SM, Kieffer TJ. Suppressing hyper-insulinemia prevents obesity but causes rapid onset of diabetes in leptin-deficient Lepob/ob mice. Mol Metab. 2016; 5: 1103-1112. doi: 10.1016/j.molmet.2016.09.007
7. Odeleye OE, de Courten M, Pettitt DJ, Ravussin E. Fasting hyperinsulinemia is a predictor of increased body weight gain and obesity in Pima Indian children. Diabetes. 1997; 46(8): 1341-1345. doi: 10.2337/diabetes.46.8.1341
8. Schonfeld P, Wojtczak L. Short- and medium-chain fatty acids in the energy metabolism – the cellular perspective. J Lipid Res. 2016; 57: 943-954. doi: 10.1194/jlr.R067629
9. Yasuaki I, Yuji K, Masataka H, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003; 422(69280): 173-176. doi: 10.1038/nature01478
10. McGarry JD, Dobbins RL. Fatty acid, lipotoxicity and insulin secretion. Diabetologia. 1999; 42: 128-138. doi: 10.1007/s001250051130
11. Gravena C, Mathias PC, Ashcroft SJH. Acute effects of fatty acids on insulin secretion from rat and human islets of Langerhans. J Endocrinol. 2002; 173: 73-80. doi: 10.1677/joe.0.1730073
12. Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest. 1994; 93(6): 2438-2446. doi: 10.1172/JCI117252
13. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle: Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963; 1: 785-789. doi: 10.1016/S0140-6736(63)91500-9
14. Randle PJ, Garland PB, Newsholme EA, Hales CN. The glucose fatty acid cycle in obesity and maturity onset diabetes mellitus. Ann NY Acad Sci. 1965; 131: 324-333. doi: 10.1111/j.1749-6632.1965.tb34800.x
15. Roden M. How free fatty acids inhibit glucose utilization in human skeletal muscle. News Physiol Sci. 2004; 19: 92-96. doi: 10.1152/nips.01459.2003
16. Roden M, Price TB, Perseghin G, et al. Mechanism of free fatty acid–induced insulin resistance in humans. J Clin Invest. 1996; 97(12): 2859-2865. doi: 10.1172/JCI118742
17. Sears B, Perry M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015; 14: 121. doi: 10.1186/s12944-015-0123-1
18. Mäkinen S, Nguyen YH, Skrobuk P, Koistinen HA. Palmitate and oleate exert differential effects on insulin signaling and glucose uptake in human skeletal muscle cells. Endocrine Connections. 2017; 6: 331-339. doi: 10.1530/EC-17-0039
19. Wolfe BM, Klein S, Peters EJ, Schmidt BF, Wolfe RR. Effect of elevated free fatty acids on glucose oxidation in normal humans. Metabolism.1988; 37(4): 323-329. doi: 10.1016/0026-0495(88)90131-X
20. Kelley DE, Mokan M, Simoneau JA, Mandarino LJ. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J Clin Invest. 1993; 92: 91-98. doi: 10.1172/JCI116603
21. Lv ZH, Ma P, Luo W, et al. Association between serum free fatty acid levels and possible related factors in patients with type 2 diabetes mellitus and acute myocardial infarction. BMC Cardiovasc Disord. 2014; 14: 159. doi: 10.1186/1471-2261-14-159
22. Cefalu WT. Insulin resistance: Cellular and clinical concepts. Exp Biol Med (Maywood). 2001; 226: 13-26. doi: 10.1177/153537020122600103
23. Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006; 444(7121): 847-853. doi: 10.1038/nature05483
24. Gagnon A, Sorisky A. The effect of glucose concentration on insulin-induced 3T3-L1 adipose cell differentiation. Obes Res. 1998; 6(2): 157-163. doi: 10.1002/j.1550-8528.1998.tb00330.x
25. Palacios-Ortega S, Varela-Guruceaga M, Martínez JA, de Miguel C, Milagro FI. Effects of high glucose on caveolin-1 and insulin signaling in 3T3-L1 adipocytes. Adipocytes. 2016; 5(1): 65-80. doi: 10.1080/21623945.2015.1122856
26. Lin Y, Berg AH, Iyengar P, et al. The hyperglycemia-induced inflammatory response in adipocytes: The role of reactive oxygen species. J Biol Chem. 2005; 280: 4617-4626. doi: 10.1074/jbc.M411863200
27. Riley CL, Bout D, Bean C, et al. UCP1 is essential for mitochondrial structural integrity and function in brown adipose tissue. FASEB J. 2017; 31(1).
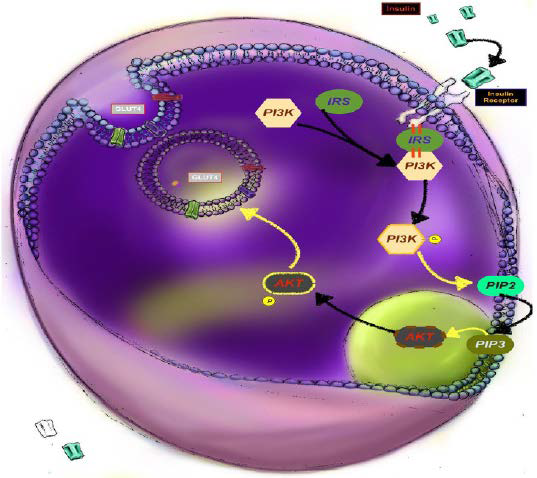
28. Rocchi S, Tartare-Deckert S, Mothe I, van Obberghen E.Identification by mutation of the tyrosine residues in the insulin receptor substrate-1 affecting association with the tyrosine phosphatase 2C and phosphatidylinositol 3-kinase. Endocrinol. 1995; 136: 5291-5297. doi: 10.1210/endo.136.12.7588273
29. Bouzakri K, Roques M, Gual P, et al. Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes. 2003; 52: 1319-1325. doi: 10.2337/diabetes.52.6.1319
30. Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988; 37(8): 1020-1024. doi: 10.2337/diabetes.37.8.1020
31. Spalding KL, Bhardwaj RD, Buchholz BA, Druid H, Frisen J. Retrospective birth dating of cells in humans. Cell. 2005; 122: 133-
143. doi: 10.1016/j.cell.2005.04.028
32. Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature. 2008; 453: 783-787. doi: 10.1038/nature06902
33. Olokoba AB, Obateru OA, Olokoba LB. Type 2 Diabetes mellitus: A review of current trends. Oman Med J. 2012; 27(4): 269-273. doi: 10.5001/omj.2012.68
34. Yin X, Lanza IR, Swain JM, Sarr MG, Nair SK, Jensen MD. Adipocyte mitochondrial function is reduced in human obesity independent of fat cell size. J Clin Endocrin Metab. 2014; 99: E209-E216. doi: 10.1210/jc.2013-3042
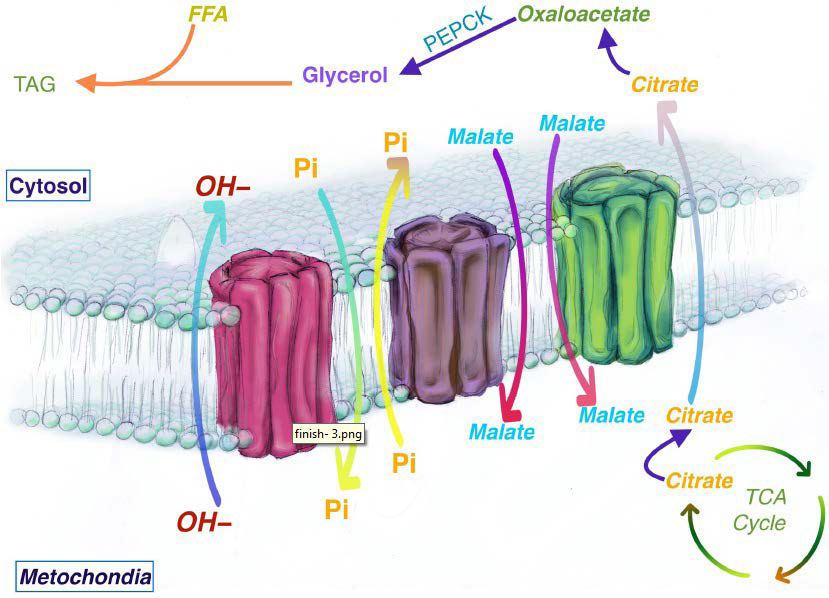
35. Hanson RW, Reshef L. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Ann Rev Biochem. 1997; 66: 581-611. doi: 10.1146/annurev.biochem.66.1.581
36. Franckhauser S, Muñoz S, Pujol A, et al. Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes. 2002; 51(3): 624-630. doi: 10.2337/diabetes.51.3.624
37. Hakimi P, Johnson MT, Yang J, et al. Phosphoenolpyruvate carboxykinase and the critical role of cataplerosis in the control of hepatic metabolism. Nutr Metab (Lond). 2005; 2: 33. doi: 10.1186/1743-7075-2-33
38. Brun T, Scarcia P, Li N, et al. Changes in mitochondrial carriers exhibit stress-specific signatures in INS-1Eβ-Cells exposed to glucose versus fatty acids. PLoS One. 2013; 8(12): e82364. doi: 10.1371/journal.pone.0082364
39. Das K, Lewis RY, Combatsiaris TP, et al. Predominant expression of the mitochondrial dicarboxylate carrier in white adipose tissue. Biochem J. 1999; 344(2): 313-320.
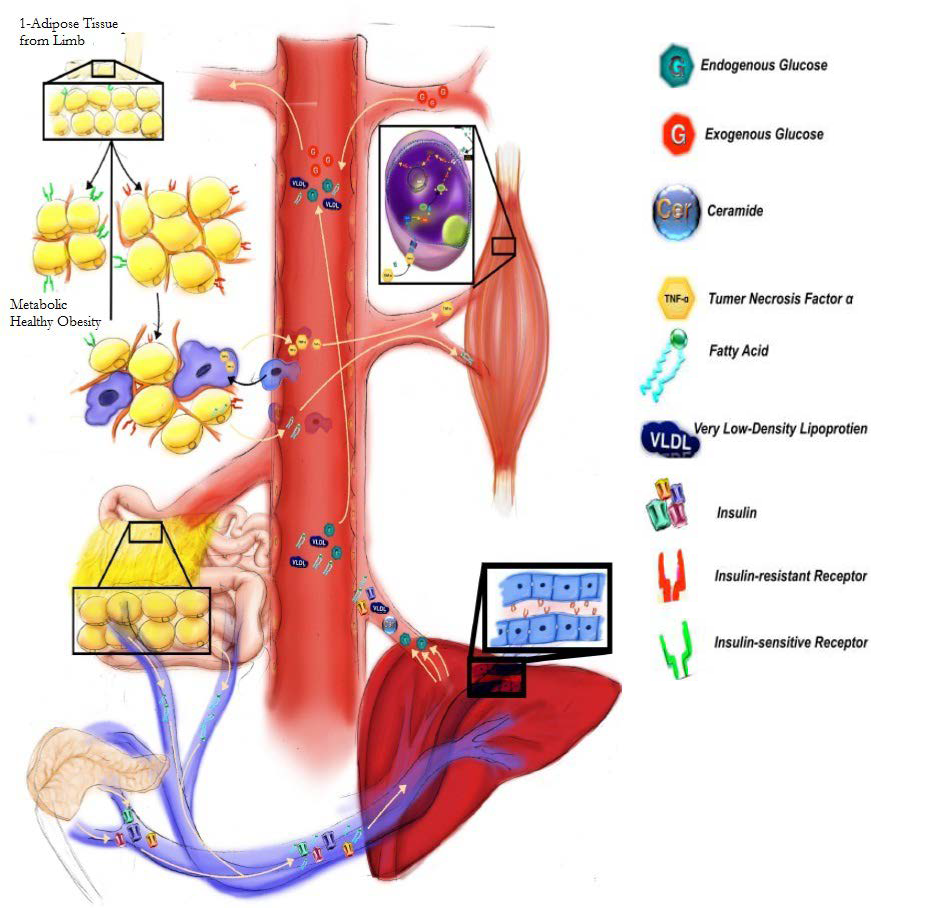
40. Arner P. The adipocyte in insulin resistance: Key molecules and the impact of the thiazolidinediones. Trends Endocrinol Metab. 2003; 14(3): 137-145. doi: 10.1016/S1043-2760(03)00024-9
41. Kim JI, Huh JY, Sohna JH, et al. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol Cell Biol. 2015; 35(10): 1686-1699. doi: 10.1128/MCB.01321-14
42. Delarue J, Magnan C. Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care. 2007; 10(2): 142-148. doi: 10.1097/MCO.0b013e328042ba90
43. Paz K, Hemi R, LeRoith R, et al. Phosphorylation of insulin receptor substrate-1 (IRS-1) by protein kinase b positively regulates irs-1 function. J Biol Chem. 1997; 272: 29911-29918. doi: 10.1074/jbc.272.47.29911
44. Liu YF, Paz K, Herschkovitz A, et al. Protein kinase c θ inhibits insulin signaling by phosphorylating irs1 at Ser1101. J Biol Chem. 2001; 276: 14459-14465. doi: 10.1074/jbc.C400186200
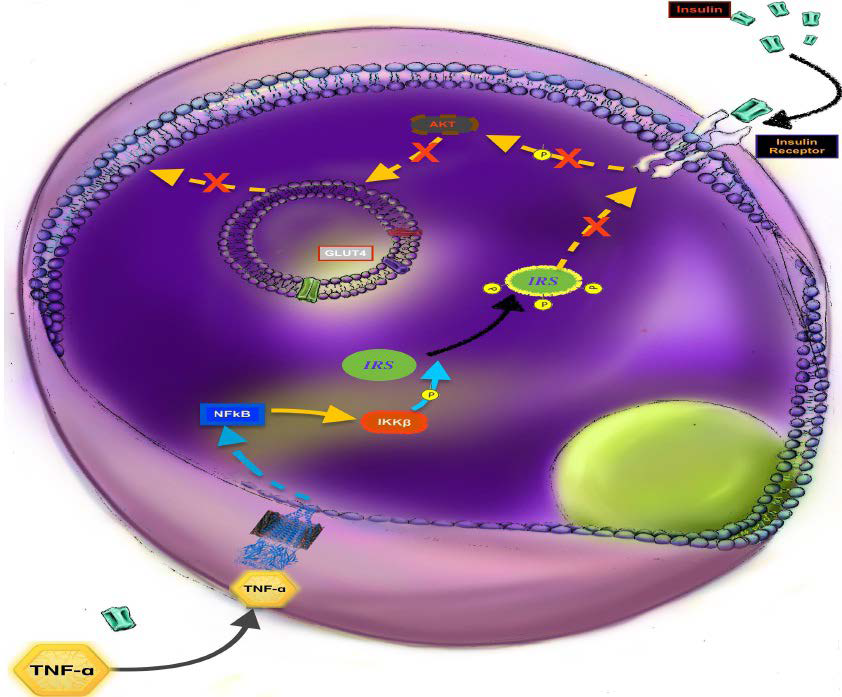
45. Ohmura E, Hosaka D, Yazawa M, et al. Association of free fatty acids (FFA) and tumor necrosis factor-alpha (TNF-alpha) and insulin-resistant metabolic disorder. Horm Metab Res. 2007; 39(3):
212-217. doi: 10.1055/s-2007-970421
46. Mlinar B, Marc J, Pfeifer M. Molecular mechanisms of insulin resistance, obesity and metabolic syndrome. Bio chemia Medica. 2006; 16(1): 8-24. doi: 10.11613/BM.2006.003
47. Le Roith D, Zick Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care. 2001; 24(3): 588-597. doi: 10.2337/diacare.24.3.588
48. Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol (Lausanne). 2016; 7: 30. doi: 10.3389/fendo.2016.00030
49. Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. J Cell Biol. 2015; 208(5): 501-512. doi: 10.1083/jcb.201409063
50. Park S-H, Liu Z, Sui Y, et al. IKKb is essential for adipocyte survival and adaptive adipose remodeling in obesity. Diabetes. 2016; 65: 1616-1629. doi: 10.2337/db15-1156
51. Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes (Lond). 2009; 33(1): 54-66. doi: 10.1038/
ijo.2008.229
52. Jiang C, Qu A, Matsubara T, et al. Disruption of hypoxia-inducible factor 1 in Adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet–fed mice. Diabetes. 2011; 60(10): 2484-2495. doi: 10.2337/db11-0174
53. Martinez-Santibañez G, Lumeng CN-K. Macrophages and the regulation of adipose tissue remodeling. Annu Rev Nutr. 2014; 34(1): 57-76. doi: 10.1146/annurev-nutr-071812-161113
54. Guzik TJ, Skiba DS, Touyz RM, Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc Res. 2017; 113(9): 1009-1023. doi: 10.1093/cvr/cvx108
55. Kang YE, Kim JM, Joung KH, et al. The roles of adipokines, pro-inflammatory cytokines, and adipose tissue macrophages in obesity-associated insulin resistance in modest obesity and early metabolic dysfunction. PLoS One. 2016; 11(4): e0154003. doi: 10.1371/journal.pone.0154003
56. Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev. 2010; 11(1): 11-18. doi: 10.1111/j.1467-789X.2009.00623.x
57. Hamdy O, Porramatikul S, Al-Ozairi E. Metabolic obesity: The paradox between visceral and subcutaneous fat. Curr Diabetes Rev. 2006; 2(4): 367-373. doi: 10.2174/1573399810602040367
58. Lafontan M. Differences between subcutaneous and visceral adipose tissues. [In:] Bastard JP, Fève B (eds). Physiology and Physiopathology of Adipose Tissue. New York City, USA: Springer. 2013. 329-349. doi: 10.1007/978-2-8178-0343-2_23
59. Andersson DP. Changes in subcutaneous fat cell volume and insulin sensitivity after weight loss. Diabietes Care. 2014; 37(7): 1831-1836. doi: 10.2337/dc13-2395
60. Preis SR, Massaro JM, Robins SJ, et al. Abdominal subcutaneous and visceral adipose tissue and insulin resistance in the Framingham heart study. Obesity (Silver Spring). 2010; 18(11): 2191-2198. doi: 10.1038/oby.2010.59
61. Fang L, Guo F, Zhou L, Stahl R, Grams J. The cell size and distribution of adipocytes from subcutaneous and visceral fat is associated with type 2 diabetes mellitus in humans. Adipocyte. 2015;
4(4): 273-279. doi: 10.1080/21623945.2015.1034920
62. Jocken JW, Blaak EE. Catecholamine-induced lipolysis in adipose tissue and skeletal muscle in obesity. Physiol Behav. 2008; 94(2): 219-230. doi: 10.1016/j.physbeh.2008.01.002
63. Thompson D, Karpe F, Lafontan M, Frayn K. Physical activity and exercise in the regulation of human adipose tissue physiology. Physiol Rev. 2012; 92(1): 157-191. doi: 10.1152/physrev.00012.2011
64. Gerhard GS, Styer AM, Strodel WE, et al. Gene expression profiling in subcutaneous, visceral, and epigastric adipose tissues of patients with extreme obesity. Int J Obes (Lond). 2014; 38(3): 371-378. doi: 10.1038/ijo.2013.152
65. Ruge T, Sukonina V, Myrnas T, et al. Lipoprotein lipase activity/mass ratio is higher in omental than in subcutaneous adipose tissue. Eur J Clin Invest. 2006; 36: 16-21. doi: 10.1111/j.1365-2362.2006.01584.x
66. Fried SK, Russell CD, Grauso NL, Brolin RE. Lipoprotein lipase regulation by insulin and glucocorticoid in subcutaneous and omental adipose tissues of obese women and men. J Clin Invest. 1993; 92: 2191-2198. doi: 10.1172/JCI116821
67. Semenkovich CF, Wims M, Noe L, Etienne J, Chan L. Insulin regulation of lipoprotein lipase activity in 3T3-L1 adipocytes is mediated at posttranscriptional and posttranslational levels. J Biol Chem. 1989; 264: 9030-9038. doi: 10.1016/S0021-9258(18)81898-1
68. Faraj M, Sniderman AD, Cianflone K. ASP enhances in situ lipoprotein lipase activity by increasing fatty acid trapping in adipocytes. J Lipid Res. 2004; 45: 657-666. doi: 10.1194/jlr.M300299-
JLR200
69. Mashek DG. Hepatic fatty acid trafficking: Multiple forks in the road. Adv Nutr. 2013; 4: 697-710. doi: 10.3945/an.113.004648
70. Kotronen A, Juurinen L, Tiikkainen M, Vehkavaara S, YkiJärvinen H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroeinterology. 2008; 135(1): 122-130. doi: 10.1053/j.gastro.2008.03.021
71. Badin P-M, Langin D, Moro C. Dynamics of skeletal muscle lipid pools. Trends Endocrinol Metab. 2013; 24(12): 589-644. doi: 10.1016/j.tem.2013.08.001
72. Shaw CS, Clark J, Wagenmakers AJ. The effect of exercise and nutrition on intramuscular fat metabolism and insulin sensitivity. Ann Rev Nut. 2010; 30(1): 13-34. doi: 10.1146/annurev. nutr.012809.104817
73. Dagenais GR, Tancredi RG, Zierler KL. Free fatty acid oxidation by forearm muscle at rest, and evidence for an intramuscular lipid pool in the human forearm. J Clin Invest. 1976; 58: 421-431. doi: 10.1172/JCI108486
74. Guo Z, Burguera B, Jensen MD. Kinetics of intramuscular triglyceride fatty acids in exercising humans. J Appl Physiol (1985). 2000; 89: 2057-2064. doi: 10.1152/jappl.2000.89.5.2057
75. Schenk S, Horowitz JF. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest. 2007; 117(6): 1690-1698. doi: 10.1172/JCI30566
76. Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab. 2001; 86(12): 5755-5761. doi: 10.1210/jcem.86.12.8075
77. Li Y, Xu S, Zhang X, Zongchun Y, Cichello S. Skeletal intramyocellular lipid metabolism and insulin resistance. Biophys Rep. 2015; 1: 90-98. doi: 10.1007/s41048-015-0013-0
78. Hansen ME, Tippett TS. Insulin increases ceramide synthesis in skeletal muscle. J Diabetes Res. 2014; 2014: 765784. doi: 10.1155/2014/765784