INTRODUCTION
Lysosomal diseases are a group of rare afflictions linked to innate errors of metabolism. Lysosomes are subcellular organelles limited by a membrane and contain many digestive enzymes involved in the catabolism of complex molecules, such as glycolipids, glycoproteins and mucopolysaccharides. The deficiency of a lysosomal enzyme obstructs the degradation of these molecules, causes their accumulation in lysosomes and creates metabolic abnormalities disrupting cell function. There are about fifty lysosomal diseases. Gaucher disease (GD) is one of the most frequent lysosomal diseases,1 with an incidence of around 1/40,000 to 1/60,000 births in the general population. The disease was first described by Philippe Gaucher in 1882 in a patient with massive splenomegaly without leukemia. Gaucher is a genetic disease caused by mutations in the GBA1 gene, located on chromosome 1 (1q21). More than 300 GBA mutations have been described in the GBA1 gene, and the most common ones are N370S, L444P, 84GG, IVS2.2 The clinical aspects of the disease are caused by a deficiency of the lysosomal enzyme, glucocerebrosidase that leads to the accumulation of its substrate (glucosylceramide) in lysosomal macrophages in the bone marrow, liver, spleen, lungs, and other organs, which contributes to pancytopenia, massive hepatosplenomegaly, and, at times, diffuse infiltrative pulmonary disease. Progressive infiltration of Gaucher cells in the bone marrow may lead to thinning of the cortex, pathologic fractures, bone pain, bony infarcts, and osteopenia. These bony features may also be related to macrophageproduced cytokines. Gaucher disease has traditionally been divided into the following 3 clinical subtypes:
Type 1–Non neuronopathic form, which is the most common type
Type 2–Acute neuronopathic form
Type 3–Chronic neuronopathic form
However, these distinctions are not absolute, and GD represents a phenotypic continuum.2
A case report of a 56-year-old patient diagnosed with GD is presented herein.
CASE DESCRIPTION
The patient was born from non Jewish consanguineous parents in 1962. She presented to the emergency department in August 2018, for extreme pain on her right hip. The pain was local without specific irradiation, increased on mobilization and improved upon taking rest. It had started one month ago, without trauma or other identified cause, and was not relieved with analgesics.
Her medical record consisted of splenectomy at the age of 9-years-old. Secondary to splenomegaly, recurrent infections in the upper respiratory system, recurrent tonsillitis, anemia that attributed to gynecological loss, fracture at L2 treated with percutaneous cimentoplasty in 2014 and an aggressive spinal hemangioma prior to that. Her sister was diagnosed with Gaucher disease at the age of 10-years-old, and she died from the disease’s complications at the age of 45.
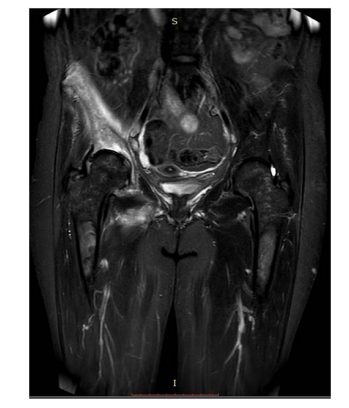
Upon admission, the blood test reports revealed the following results: Hb 11.5 g/dL, MCV 95 fl, white blood cells 8200/ mm3, platelets 261000/mm3, ferritin 2654 ng/mL, normal liver function tests, and normal creatinine clearance. Right femoral Xray revealed an Erlenmeyer flask sign (Figure 1). The magnetic resonance imaging (MRI) of the right hip revealed diffused significant heterogeneity in the signal of the trabecular bone of pelvic bones, femur and the visualized part of the lumbar spine, with individualization in particular of a much more diffuse and more intense signal abnormality in the spongy bone of the right iliac bone involving the acetabulum and the ilio and ischio-pubic branches, with cortical thinning visible at the height of the iliac crest, and a very important edema of the adjacent soft parts, involving the muscles iliopsoas, glutes, adductors and external obturators on the right side (Figure 2). The MRI concluded no osteonecrosis of the iliac bone, acetabulum and the femoral head.
Figure 1. Erlenmeyer Flask Sign of the Femur
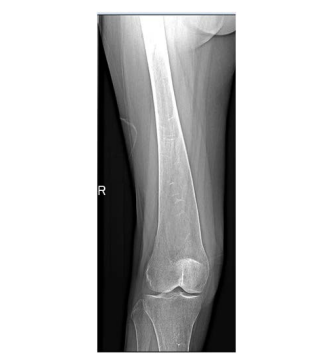
Figure 2. Diffuse Significant Heterogeneity in the Signal of the Trabecular
Bone of Pelvic Bones, Femur
Taking into consideration the clinical presentation of the patient (splenomegaly, hepatomegaly confirmed by abdominal ultrasound, anemia, high ferritinemia, bone lesions) and the family history, the patient was suspected to have Gaucher disease.
In order to confirm the diagnosis, the dosage of the lysosomal enzymes from dried blood, and the molecular genetic testing were performed. The first test revealed that the activity of betaglucosidase was below its reference range (24.06 pmol/spot*20 h), with an elevated concentration of glucosylsphingosine, and the later tests showed a homozygous mutation of p.V414G which is similar to the known missense mutation p.V414.
The patient was operated in January 2019 with a total right hip replacement and was treated, since November 2018, with intra-venous Imiglucerase (60 UI per kilogram) once every 2-weeks. Moreover, she presented two fractures on D11 and D12, confirmed with lumbar MRI, in March 2019.
DISCUSSION AND CONCLUSION
Although it is the most common all the lysosomal storage diseases, Gaucher disease still remains rare and most cases present a gradual onset phenotype, which explains its delayed diagnosis. The spectrum of the disease is wide, and its clinical manifestation varies from being asymptomatic to presented with all complications. This patient had presented splenomegaly at infancy, which led to splenectomy at the age of nine, without any diagnosis. It is important to include Gaucher disease in the diagnostic decision tree in cases of splenomegaly and/or thrombocytopenia, in order to avoid potentially harmful splenectomy. Before the advent of enzyme replacement therapy (ERT), total or partial splenectomy was the standard of care for massive splenomegaly or severe pancytopenia.3
Another common manifestation of GD is a bone disease. The bone lesions in Gaucher disease are caused mainly due to alterations in its metabolism (turnover, remodeling, and mineralization), leading to various skeletal complications such as osteoporosis, marrow infiltration, avascular necrosis or osteolysis.4 Patients with preexistent skeletal complications tend to suffer episodes during ERT, such as medullary infarctions, avascular necrosis, or fractures, but the frequency of these occurings is reduced.3 In our case, the patient had aseptic necrosis of the right femoral head and iliac bone that was treated surgically with total hip replacement shortly after being started on ERT.
The efficacy of ERT on hematological and visceral parameters, such as anemia, thrombocytopenia and splenomegaly, is well established in the literature.5 However, enzyme therapy cannot reverse established osseous injury; pathological damage such as osteonecrosis, bone infarcts and fracture, once it has occurred, is irreversible. Thereby, despite the low risk of early failure due to aseptic loosening, total hip replacement in patients with Gaucher’s disease with symptomatic osteonecrosis of the femoral head is advised.4
On an additional matter, patients with GD suffer from early osteopenia. The bone mineral density tends to increase during ERT but with a slow rate, hence early diagnosis and treatment becomes important, in order to avoid severe complications. Furthermore, the use of bisphosphonates can be an effective and safe mean to increase bone density.
CONSENT
The authors have received written informed consent from the patient. CONFLICT OF INTEREST The authors declare that they have no conflicts of interest.


