INTRODUCTION
Medical devices (MDs) include a wide variety of products, ranging from a simple plaster to a cardiac stent, or a laser eye scan. The importance of these instruments in the healthcare industry is remarkable; in fact, the European Union (EU) market of MDs represent 25% of the global market, with a turnover of about 100 billion Euros, and with more than 500,000 employees.
In May 2017, the European Parliament issued Regulation 2017/745 (Reg. 2017/745). The latter, introduced several major changes in planning, conducting and reporting of clinical investigations (CIs), which will be expected to come into effect within a transition period of three years following its approval. Manufacturers and independent researchers will have to adapt quickly to this complex new regulation, due to the relatively short transition period. The aim of the present work is to demonstrate an overview which will serve as a reference to the researchers willing to plan and conduct a CI according to the new regulation.
REGULATORY FRAMEWORK
Regulation 2017/745 replaced the Directives 93/42EEC on MDs and 90/385 EC on the active implantable medical device (AIMD). The aim of the new regulation was to ensure better facilities of patient protection, to achieve higher quality standards for MDs, to promote cooperation between EU member states (MS) and to harmonize the procedures across the EU. Reg. 2017/745 imposed a major change in the domain of MDs: products previously regulated as commercial goods were expected to fall under Reg. 2017/745 (e.g., cosmetic contact lenses) while some existing MDs were assigned to a different risk class. Moreover, additional rules for the classification of MDs were introduced, and other changes were implicated in the notified bodies (NBs) and conformity procedures. Also, the CIs were associated with these changes.
A comprehensive analysis of Reg. 2017/745 and other references on CIs would probably require a textbook. Therefore, six main points of direct practical relevance for professionals involved in design and conducting a CI have been discussed: (1) role of CIs in the context of Clinical Evaluation of an MD, (2) objectives of CIs, (3) cases where a CI is mandatory and their exceptions; (4) application procedure for a CI; (5) requirements for conducting a CI, (6) serious adverse events reports, (7) reporting the results of CI. In this review, a summary following the analysis of relevant articles of Reg. 2017/745 have been presented, acting as a reference guide to researchers willing to undertake a CI.
SOURCES
The European Regulation 2017/745 has been consulted as the main source of information for the present study.1 CIs have been discussed mainly in its Chapter VI and Annex XV. Directive 93/42 CE2 as amended by directive 47/20073 was also consulted which has been referred to as medical device directive (MDD), and the International Organization for Standardization (ISO) standard 14155:2012,4 which outlines the technical guideline for the designing, conducting, analysis and reporting of CIs. Additional sources of information available include MEDDEV2.7/15 on clinical evaluation, 2.7/26 assessment of CI application, 2.7/37 reporting of serious adverse events, 2.7/48 on CIs. For an understanding of the regulations and guidelines, the websites of European Commission9 and Italian Health Ministry10 were also consulted. For economic data recorded from the MD industry, the website of the European Commission (EC)11 and the report of the Italian Association of Medical Device Manufacturers Assobiomedica (2016)12 served as the relevant resources.
THE CLINICAL INVESTIGATION, REGULATORY FRAMEWORK
Role of CIs in the context of the Clinical Evaluation of MD
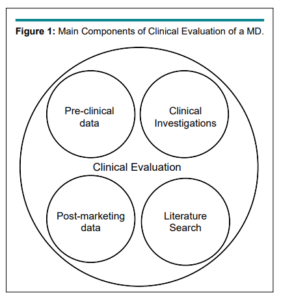
Reg. 2017/745 defined CI as “any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device”. CIs are the key components of the wider clinical evaluation process of an MD. The clinical evaluation of MDs are a comprehensive process of assessment, intended towards verifying whether the MD meets the essential requisites defined in Annex I of Reg. 2017/745. The Clinical Evaluation is based on: a) MD technical data (projects, materials, etc.); b) preclinical data (usually the tests described in ISO 10993 guidelines) and c) clinical data. CIs are considered as the most relevant source of clinical data to evaluate an MD. Reg. 2017/745 dealt with clinical evaluation in chapter VI, Article 61 and Annex XIV, while MEDDEV 2.7/1 Rev.4 (2016) provided a detailed guide for clinical evaluation.
Figure 1 shows the main stages of clinical evaluation. The clinical evaluation process continued following the CE mark of the device, and CIs were still required even when the MDs were marketed (post-marketing CIs). Thus, it could be concluded that each CI should be planned in view of its use in the wider clinical evaluation of the MD.
Aims of Clinical Investigations
Article 62 of Reg. 2017/745 lists the aim of CIs. The latter must assess that, under the normal conditions of use, a particular MD: (a) reached the intended purpose planned in its design and works as expected; and/or (b) provided the expected clinical benefit; and/or (c) showed an acceptable risk/benefit ratio, considering the side effects weighed against the benefits to be achieved by the device. Each CI can be indicative of compliance with one or more of the above-mentioned parameters; however, the possible aims were limited to the list above for pre-marketing and postmarketing CIs.
CIs should be designed bearing in mind that in these kind of investigations, there is little room for speculative research. Pre- and post-marketing CIs should be aimed at generating data needed for clinical evaluation or post-marketing clinical follow-up (PMCF).
Cases where Clinical Investigations are Mandatory and their
Exceptions
In Article 61, Reg. 2017/745 stated that CIs are mandatory for all implantable and class III devices. CIs are not mandatory if a new implantable or class III device is equivalent to a device already marketed by the same manufacturer and the clinical evaluation of the latter is sufficient to demonstrate the conformity of the new one. CIs are not mandatory for class III devices already marketed under MDD and for implantable devices like sutures, staples, tooth crowns (see full list that has been mentioned in the paragraph 6 of the Article 61), that have sufficient clinical data to demonstrate their conformity. It is important to note that, in the absence of clinical data, a low-risk MD requires a CI. Finally, according to the characteristics of the device, and including low-risk device, post-marketing CI are requested to confirm, integrate or update existing clinical data, especially to address the safety concerns of the device within the frame of a PMCF.
Application and Assessment of a Clinical Investigation
Before Reg. 2017/745, the application for a CI was mainly a national procedure; therefore, the documents to be submitted, the requirements and the time limits for decision could be different between member states (MS). According to Reg. 2017/745, the application must be submitted to the MS where the CI should be conducted (concerned MS), via institutional EU-wide internet portal. The documents to be presented will be standardized for all MS, and are listed in Appendix XIV of the regulation. A single, unique identification number, valid in the EU, will identify the CI, to allow easier tracking of the CI itself. For class III implantable, and class IIb active devices intended to administer and/or remove a medicinal product, the sponsor (both Manufacturer and independent researcher) may request for the opinion of a group of EU-appointed experts. This scrutiny procedure is particularly useful for more complex and innovative MDs. Interestingly, the CI for class I, IIa and IIb MDs, may start immediately after the validation of the application, unless there be a negative opinion from the National Ethics Committee (NEC).
Within 10 days after receiving the application, the concerned MS will confirm the receipt of the application dossier and its completeness. The confirmation date is considered as the validation date. The concerned MS shall notify the sponsor of the authorization within 45 days after the validation date. If during the assessment process the concerned MS needs additional information, it the may request additional information from the sponsor. The required time for question and answer will not be counted within the 45 days (clock-stop). The concerned MS may postpone the authorization of the CI for a further 20 days to allow for consultation with the experts.
For multinational studies, the sponsor proposes in the application that one of the MSs where the CI should be conducted acts as a coordinating Member State. Within six days from the application, MSs may agree with the sponsor’s proposal or can decide that a different authority will function as the coordinator. In case of disagreement between the MSs, the CI will be coordinated by the MS proposed by the sponsor. The coordinating MS will issue and distribute the CI draft assessment to the other MSs within 26 days. By day 38, MSs will notify of their comment, and by day 45, the final assessment report will have to be transmitted to the sponsor. Each MS has, however the right to request additional documentation once, and to disagree with the conclusion of the coordinating MS. The coordinating MS may extend the evaluation of class IIb and class III devices for another 50 days. It is believed that this procedure allows for a faster authorization of multinational CI while ensuring better quality services for patient protection.
Conduct of a Clinical Investigation
Guidance and guidelines: Reg. 2017/745 outlines the general recommendations for conducting CIs (Article 72). Detailed guidance is given in accordance with the quality standards of ISO 14155:2012,4 and guidelines MEDDEV2.7/15 (clinical evaluation) 2.7/26 (assessment of clinical investigations) 2.7/37 (adverse event reporting) 2.7/48 (clinical investigations). Issued by the EC, MEDDEV are not legally binding; however, they report the EC interpretation of regulations, and therefore, any deviation from the MEDDEV should be justified. Guideline IMDRF/MC/ N25FINAL:201513 issued by the International Medical Device Regulators Forum (IMDRF) guides the implementation of ISO 14155: 2011.
Protection of Patients: Reg. 2017/745 is associated with seven Articles (62 to 69) dedicated to patient protection. Detailed instructions have been laid down for; informed consent (Article 63), CI, on incapacitated subjects (Article 64), minors (Article 65), pregnant and breast-feeding women (Article 66). Moreover, Reg.2017/745 allows MS to define additional national measures (Article 67) and establish rules to ensure adequate compensation for damage (Article 69). The primary objective is to satisfy individual patient interest rather than focus on the broader and vague concept of promoting “interest of community”. Practically, CI should directly benefit the individual subjects enrolled in the study, while avoiding any unnecessary and minimizing any inevitable discomfort to the patients. In accordance with the stated regulations, economic incentives are forbidden for minors, incapacitated subjects, pregnant and breast-feeding women (with the exception of reimbursement of expenses) in order to prevent the economic exploitation of the potential subjects. In conclusion, when planning for a CI, it is important to consider the issues related to the patient’s safety and review carefully all the issues concerning the populations involved.
Serious Adverse Event (SAE)
According to Reg. 2017/745, serious adverse events (SAE), device deficiency that could lead/could have led to a SAE, and its follow-up must be reported via the internet portal defined in Article 73. SAE includes AE that could lead to death, serious conditions of ill-health of the subject, foetal distress and/or death, congenital anomaly or birth defect. MEDDEV 2.7/3 Rev 3 defines device deficiency as “Inadequacy of an investigational medical device related to its identity, quality, durability, reliability, safety or performance. This may include malfunctions, error in use, or inadequacy in the information supplied by the manufacturer”. It is important to note that any form of deficiency that could have led to a SAE should be reported as if the SAE occurred. SAEs must be notified within strict timelines: the investigator must report the occurrence of the above-mentioned events to the sponsor no later than three days after the event occurred. Conversely, the sponsor must report any of the above events to the concerned MS, within two days in case of an event that may have caused death or posed an imminent risk of death or injury requiring immediate action, and within seven days for other events. MEDDEV 2.7/3 Rev. 3 provides the form for the reporting of SAEs and deficiencies.
Reporting of a Clinical Investigation
The sponsor must inform the concerned MS with in 15 days when the CI is concluded. Irrespective of the CI outcome, the sponsor shall submit the clinical investigation report and a summary presented in terms easily understandable to the intended user, by means of the electronic system referred to in Article 73. The clinical investigation report and the summary must be submitted within one year of the end of the CI or within three months of the early termination or temporary halt. The clinical investigation report and the summary will be accessible to the public as soon as the device receives the CE mark. If, due to scientific reasons, the clinical investigation report is unavailable for one year, it is advisable to provide a preliminary justification in the clinical investigation plan. The CI report will include a summary of the clinical investigation plan, the results of the clinical investigation, the summary of adverse events and deficiencies and finally, the discussion and overall conclusions.
DISCUSSION AND CONCLUSION
Reg. 2017/745 leads to a major transformation in the field of MDs, including the CIs. Planning, design, conducting and reporting a CI must be documented and disclosed to the competent authorities in accordance with the new regulations. In addition, it is of primary importance that each CI is considered as an aspect of a formal procedure that begins with the conformity procedure (CE marking) and continues for the entire life of the device. Evaluation parameters and format of data collected should allow for pooling of information and future meta-analysis. In the light of the information presented in this study, it is of great importance to emphasize that designing, implementing and conducting of CIs demand that the personnel in charge have received the requisite training and expertise in the field of MD.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.


