INTRODUCTION
The relationship between cardiac and hepatic dysfunction has been a well-recognized entity for over two centuries.1 Yet, the complexity and nuances of the association still remain a topic of intense interest and research. Studies dealing with this topic are relatively few, not rarely with contradictory results. There are several reasons for the variant results: heart failure etiology has changed over the years, being mainly related to rheumatic valvular disease in the earliest studies and to ischemic cardiomyopathy more recently.2 Also, the outcome of heart failure has dramatically improved due to superior medical therapies, not to mention widespread use of heart transplantation. Thus, cardiac cirrhosis, once the paradigm of liver involvement in heart failure, is now rare.
Concomitant hepatic and cardiac disorders may be categorized according to etiology. That is: (i) cardiac disease affecting the liver, (ii) hepatic disease affecting the heart, or (iii) cardiac and hepatic disease secondary to a shared etiology.3,4 In this review, we chose to focus on “cardiac hepatopathy”, or hepatic pathology secondary to cardiac dysfunction.2 As will be described herein, “cardiac hepatopathy” includes a spectrum of altered clinical, biochemical, histological, and hemodynamic disturbances. It is classically described in the setting of either acute or chronic heart failure. However, clinical and pathogenetic factors related to both conditions often co-exist.
MACRO- AND MICROCIRCULATION OF THE LIVER
In order to understand the range of hepatic abnormalities that characterize cardiac hepatopathy, it is important to first appreciate the unique anatomy and physiology of the liver.
Macrocirculation
The liver has a rich dual blood supply derived from both the portal and systemic vascular compartments: the portal vein supplies two thirds of hepatic blood flow and the hepatic artery is responsible for the remaining third.5 Although the blood supply from the portal vein is less oxygenated compared to the hepatic artery, the portal vein supply is full of nutrients as it drains the vascular beds of the stomach, intestine, and spleen. An understanding of how the liver regulates its dual blood supply is also critical, especially with respect to compensatory mechanisms in the face of hemodynamic compromise. In order to maintain constant sinusoidal pressure to the hepatic beds, the liver employs an autoregulatory mechanism whereby a decrease in blood flow via the portal vein is matched by a compensatory dilation of the hepatic artery and thus increased flow and maintenance of perfusion.5 However, the opposite does not hold in that a decrease in hepatic arterial blood flow that occurs secondary to a reduced cardiac output in left heart failure is not matched by a compensatory increase in portal venous inflow.1,6,7
Microcirculation
Liver architecture has been traditionally described in terms of the histological unit and the functional unit. The histological (or “classical”) unit of the liver is the lobule, while the functional unit of the liver is the acinus.8 The classical lobule is hexagonal in shape, bounded by the portal triads with the central vein at the center, and can be divided into concentric, centrilobular, midzonal, and periportal parts. The acinus is diamond-shaped and has at its center a line connecting two portal triads. The acinus is divided into zone 1 (periportal), zone 2 (transition), and zone 3 (centrilobular) according to the direction of flow of oxygen- and nutrient-rich blood from zone 1 closest to the portal triad to zone 3 surrounding the terminal hepatic vein.9
CONGESTIVE HEPATOPATHY
Congestive hepatopathy refers to the spectrum of chronic liver injury attributed to passive hepatic congestion arising in the setting of right-sided heart failure or any cause of increased central venous pressure,10 including biventricular failure from cardiomyopathy,11 severe pulmonary hypertension or cor pulmonale,12 constrictive pericarditis13 as well as valvulopathies such as mitral stenosis14 and tricuspid regurgitation. This condition was first described by Dame Sheila Sherlock in her seminal work on the topic in 1951.1
Histopathology and Pathogenesis
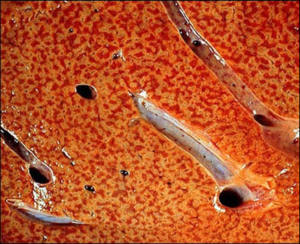
On gross examination, the congested liver is an enlarged, purple-hued organ with prominent hepatic veins. The cut surface conforms to the classic “nutmeg” appearance, reflecting the alternating pattern of hemorrhage and necrosis of zone 3 (red) with normal or slightly steatotic areas in zones 1 and 2 (yellow) (Figure 1).
Figure 1: Cut surface of the congested liver reminiscent of the classic “nutmeg appearance” which is caused by chronic passive congestion of the central veins with hemorrhage and necrosis in zone 3. Red cells pool and distend the sinusoids around the central vein. These regions
develop a darker red-violet color, in contrast to the surrounding tan liver parenchyma.
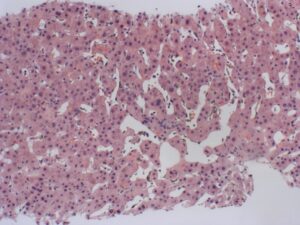
In the face of decreased perfusion, zone 1 hepatocytes are the least susceptible to necrosis, while zone 3 hepatocytes are the most susceptible. Furthermore, zone 3 hepatocytes are the most susceptible to damage induced by passive congestion secondary to right heart failure (Figure 2). Hepatic sinusoids lack a basement membrane and have a characteristic fenestrated, discontinuous endothelial lining that also contains macrophages specific to the liver (Kupffer cells). The hepatocytes themselves are separated from the sinusoids by an interstitial space, the Space of Disse. Under normal physiologic conditions, free flow through the sinusoidal fenestrations ensures a low hydrostatic pressure.15 With passive congestion of the liver in right heart failure, the increased hydrostatic pressure produces sinusoidal edema and hemorrhage, which eventually compromises oxygenation.
Although the pathogenesis of fibrosis in cardiac hepatopathy is relatively well-characterized, it still remains unclear as to why some cardiac patients develop hepatic pathology and others do not, as the stage of congestive heart failure does not seem to correlate well with hepatic fibrosis and cirrhosis.1,2,15,16,17 Prolonged or repeated episodes of hepatic congestion with fibrosis may very rarely lead to so-called “cardiac cirrhosis”. It must be noted, however, that the entity of cardiac cirrhosis, also referred to as congestive cirrhosis, is somewhat elusive, with some authors not considering it true cirrhosis.18 Uniquely, the fibrosis of cardiac hepatopathy is predominantly around the central hepatic veins with relative sparing of the portal tracts (“reverse lobulation”), although extension is possible with repeated attacks.1,18,19 This is distinct from other etiologies of cirrhosis, in which fibrosis generally occurs first in the area around the portal tract.
Figure 2. H/E section of liver (10x magnification) with sinusoidal dilation in zone 3. As the severity of the lesion increases, the sinusoids around the central vein become distended with extravasated red cells and there is adjacent hepatocyte plate atrophy.
Incidence
The incidence of congestive hepatopathy, significant fibrosis or cardiac cirrhosis ranges between 15% to 65% of patients with significant heart failure.2,20,21 By today’s accounts cardiac cirrhosis is rare. In a study by Myers et al.2 of 83 subjects with heart failure, significant fibrosis with architectural distortion was found in only 19% of cases with only one individual having an established diagnosis of cirrhosis. In a smaller series of 59 patients awaiting cardiac transplant or Left-ventricular assist device (LVAD) placement, congestive changes were seen universally with 19% having histologic changes consistent with cirrhosis.22
Clinical Features
In the majority of patients, the clinical picture is dominated by signs and symptoms of right-sided heart failure rather than those of liver disease (Table 1). Hepatomegaly is the most common manifestation with reports as high as 95% to 99% in acute or chronic heart failure. A mild, dull, right upper quadrant pain is often present and is likely secondary to hepatomegaly and stretching of Glisson’s capsule. An additional physical finding includes a pulsatile liver, which essentially results from volume overload of the right atrium.23 Importantly, loss of the pulsatile liver in chronic cardiac disease is more concerning than its positive presence,23,24 as loss of pulsatility implies progression to cardiac fibrosis or cirrhosis and warrants attention.
Table 1: Congestive Hepatopathy: Signs and symptoms
|
Symptom/Sign
|
Patients with acute or chronic heart failure showing sign/symptom (% range)
|
|
Hepatomegaly
|
95-99% |
| Marked hepatomegaly (> 5cm below right costal margin) |
49-57%
|
|
Peripheral edema
|
71-77% |
| Pleural effusion |
17-25%
|
|
Ascites
|
7-20% |
| Splenomegaly |
20-22%
|
|
Jaundice
|
10-20%
|
Other common, yet nonspecific, findings include peripheral edema, pleural effusion, splenomegaly, and jaundice (Table 1). Ascites is also clinically present in up to 20% of patients with congestive hepatopathy (although 41% at autopsy have ascites).25 However, it must be noted that the ascites is a result of right-sided heart failure and not intrinsic liver dysfunction, as is the case in other causes of cirrhosis. Although the Serum ascites-albumin gradient (SAAG) is greater than 1.1 g/dL, consistent with portal hypertension, the ascitic fluid protein level is characteristically high, oftentimes >2.5 g/dL. This high protein content is an indication of the preserved synthetic function of the liver,26 a finding unique to cardiac cirrhosis and critical in differentiating it from other causes of cirrhosis. The underlying pathophysiology of cardiac ascites remains uncertain, but some have proposed that sinusoidal hypertension with disruption of fenestrae ultimately allows for exudation of a protein rich fluid.6,26 Other useful ascitic fluid parameters are the Lactate dehydrogenase (LDH), and red cell counts, as these generally tend to be higher in cardiac cirrhosis.26
Congestive changes can readily be seen on abdominal imaging. Liver ultrasonography typically shows hepatomegaly with a homogeneous increase in echogenicity throughout the liver and dilation of the suprahepatic veins and Inferior Vena Cava (IVC). Computed tomography and magnetic resonance imaging will similarly demonstrate hepatomegaly, distension of the hepatic veins and IVC, early reflux of contrast material from the right atrium to the IVC, and a heterogeneous, mottled-appearing liver parenchyma, often referred to as a mosaic pattern, which corresponds to the nutmeg liver seen on gross inspection.27 Ascites, pleural and pericardial effusions are also frequently reported.
In terms of hemodynamic parameters, heart failure patients exhibit an increased right atrial pressure and the free and wedged hepatic venous pressures are also commonly elevated, with a normal hepatic venous pressure gradient.28,29 This finding of normal intrahepatic portal pressures is clinically relevant and likely underlies, at least in part, the minimal hepatic symptomatology associated with the majority of cases of cardiac hepatopathy.
Histologically, relative sparing of the portal tracts from fibrosis – a distinguishing factor of cardiac cirrhosis compared to other etiologies of cirrhosis as previously noted – also likely contributes to the lack of stigmata classically associated with portal hypertension. Spider angiomata and portosystemic shunts like hemorrhoidal plexus varices, caput medusae, and esophageal varices are very rarely, if at all, present in cases of cardiac hepatopathy.6,15,30 Even with progression to cirrhosis, hepatic symptoms and manifestations of portal hypertension do not predominate, which is again in contrast to cirrhosis of other etiologies.
Congestive heart failure results in a broad range of liver biochemical abnormalities. Generally, a hepatocellular pattern with predominantly elevated transaminases is seen in hypoxic hepatitis, which is a rare occurrence given that the liver is relatively protected from hypoperfusion and hypoxia. More contemporary research describes the biochemical profile in congestive heart failure as mostly cholestatic. In their study in the 1960s, Richman et al. correlated alterations in Liver Function Tests (LFTs) with either acute or chronic decompensated right-sided heart failure regardless of etiology or severity of heart disease.25 In acute dysfunction, both excretory function and parenchymal destruction were most pronounced, while a cholestatic pattern was most pronounced in chronic decompensation, findings that have been corroborated in a more recent study by Myers et al.2
Elevated serum bilirubin is also a common finding in cardiac hepatopathy, except perhaps in constrictive pericarditis,31,32 with reports of mild elevation (usually <3 mg/dL and mostly unconjugated) in up to 70% of patients.6,15,25 The hyperbilirubinemia in congestive heart failure is multifactorial and likely results from a combination of hepatocellular dysfunction, obstruction secondary to passive congestion and pressure atrophy of the canaliculi, pulmonary infarction, bile thrombi, hemolysis, and medications.1,31 Increases in the serum bilirubin have been shown to correlate with the severity of right atrial pressure and passive congestion.1,33 Bilirubin is significantly more elevated in patients with physical exam findings of volume overload, such as S3 gallop or pulmonary crackles, thus implicating its value as a prognostic factor and indicator of more severe hemodynamic dysfunction.4 Despite the common finding of hyperbilirubinemia, the presence of clinical jaundice is not common.
Decreased albumin (seen in about 30-50% of cases) is a very nonspecific finding, as it is overwhelmingly common in hospitalized patients and those with chronic diseases. With respect to synthetic function, Prothrombin Time (PT) may be more useful than albumin level at tracking progression of cardiac hepatopathy based on the observation that PT fails to correct with Vitamin K34 but does usually correct with compensation of heart failure, suggesting a direct effect on hepatic synthesis. The PT is mildly abnormal in 80% of cases. Although serum ammonia level is occasionally increased, hepatic encephalopathy is not a salient feature of congestive heart failure.35
Treatment
The cornerstone of management of all forms of congestive hepatopathy, from asymptomatic, mild elevations in hepatic indices to cardiac cirrhosis is targeted toward treating the underlying cardiac dysfunction and any triggers accounting for acute decompensation. Reversibility of biochemical aberrations in cardiac hepatopathy was described as early as 1930 when Jolliffe et al. reported normalization of liver biochemistries with restoration of appropriate cardiac function.24
Jaundice, hepatic congestion and ascites may respond dramatically to therapy with diuretics; however these drugs should be used with caution to avoid dehydration, hypotension and hepatic ischemia by precipitating zone 3 necrosis.36 It is of vital importance to maintain an adequate cardiac output. Serial large-volume paracenteses can relieve symptoms in those with diuretic-refractory tense cardiac ascites6,37,38 but over time can lead to protein loss and exacerbate the protein malnutrition commonly seen in those with advanced heart failure. Transjugular Intrahepatic Portosystemic Shunts (TIPS) or peritoneal-venous shunts are contra-indicated in this population as they can lead to exacerbation of the underlying heart failure. Cautious use of anticoagulants is advised because patients have a baseline mild increase in PT/INR and are especially sensitive to warfarin and other related compounds.39 In patients refractory to medical therapy who are suitable operative candidates, both LVAD implantation40,41 and cardiac transplantation42 have been shown to improve and reverse the congestive liver injury associated with the failing heart. In select patients with established cirrhosis, combined heart and liver transplant is a feasible option.43 Recently, there has been a report of possible reversal of cardiac cirrhosis with heart transplantation alone, effectively removing the source of the insult.44 However, such cases are the exception.
Prognosis
Over time, hepatic function typically remains stable and even when cardiac cirrhosis and ascites ensue, patients with congestive hepatopathy rarely develop other features of hepatic insufficiency.1 Fulminant liver failure, although documented, seems to be restricted to those with superimposed ischemic liver injury rather than passive congestion alone.45,46
Several studies have addressed the prognostic importance of liver function abnormalities in predicting short and long-term outcomes. According to the CHARM investigators, abnormal levels of total bilirubin, direct bilirubin, alkaline phosphatase, and albumin are statistically significant prognosticators of outcome Total bilirubin was reportedly more predictive of adverse prognosis than even the New York Heart Association functional class, left ventricular ejection fraction, diabetes mellitus, and serum creatinine.20,47 Batin et al demonstrated that the greatest prognosticators in chronic heart failure were AST and total bilirubin;48 while in a Japanese chronic heart failure study, total bilirubin, alkaline phosphatase and GGT levels were all associated with worsened outcomes.49
HYPOXIC HEPATITIS
Hypoxic hepatitis is defined as an acute and reversible significant elevation of serum AST and ALT levels to more than 20 times the upper limit of normal in the absence of known acute hepatitis or hepatocellular injury and with an appropriate clinical picture specifically involving acute circulatory, cardiac, or respiratory failure.50
More recent literature has proposed hypoxic hepatitis as a more appropriate name than “shock liver”51 and/or “ischemic hepatitis”52 given that, regardless of etiology (cardiogenic or otherwise), the underlying mechanism appears to be hypoxia even in the absence of ischemia.34 Classically, “ischemic hepatitis” was used because of the histological appearance of centrilobular necrosis, loss of hepatocytes, and sinusoidal congestion with erythrocyte extravasation, but a characteristically unremarkable inflammatory infiltrate.34,53 Although centrilobular necrosis is a critical part of the disease, histologic confirmation is seldom obtained.50,53
Incidence
Because of increased awareness and recognition of the possibility of hypoxic hepatitis accounting for elevated aminotransferases, it is now identified as the most common cause of acute liver injury , even exceeding drug-induced liver injury and acute viral hepatitis.53,54 It is well-reported that the incidence is highest in cardiac care and surgical intensive care units, with some reports identifying up to 22% of patients55 compared to a recently reported 11% in medical intensive care units50 and less than 1% incidence in the non-critical care units.56
Histopathology and Etiopathogenesis
Cardiovascular disease is recognized as the most common cause of hypoxic hepatitis, underlying over 70% of cases, while the remaining 30% of cases are split equally between respiratory failure and sepsis.57,34 The association of heart disease with increased proclivity toward developing hypoxic hepatitis might stem from passive congestion of the liver compromising its relative resistance to ischemia and hypoxia. The liver is normally well-equipped to compensate for and withstand hemodynamic derangements as evidenced by the low incidence of hepatic damage in the face of shock and circulatory collapse. However, the compensatory mechanisms are notably overwhelmed in the face of persistent hypotension or severe hypoxemia and underlying cardiac dysfunction. Virtually any cause of shock or hemodynamic instability can result in ischemic injury to the liver (See Table 2 for a complete list of causes).
Table 2: Causes of Hypoxic Hepatitis
| Heart Failure with or without cardiogenic shock |
| – Right ventricular failure
– Right ventricular myocardial infarction
– Pulmonary embolism
– Cor pulmonale
– Primary pulmonary hypertension |
| Hypovolemic shock
– Hemorrhage
– Dehydration
– Major burns |
| Other systemic disorders
– Major trauma (crush injury)
– Sepsis
– Heat stroke
– Vasculitis |
Rare causes
– Sickle cell crisis
– Carbon monoxide poisoning
– Dissecting aortic aneurysm
– Hepatic artery occlusion in the setting of a liver transplantation or with pre-existing portal vein thrombosis |
Clinical Features
Patients with hypoxic hepatitis tend to be older, predominantly male and acutely ill in the intensive care unit.55 Signs and symptoms of acute liver injury are usually absent, in contrast to other causes of acute liver injury. The clinical presentation is usually consistent with cardiac compromise53,57 although some individual studies have reported a variety of other symptoms, ranging from predominantly gastrointestinal with nausea, vomiting and diarrhea58 to an encephalopathic picture with altered mental status or even coma.34,59 The latter picture of acute fulminant hepatic failure, although rare, is more likely to occur in the presence of underlying congestive heart failure or cirrhosis.34 There are no unique physical examination findings although some patients exhibit right upper quadrant tenderness to palpation.
Despite the potential nonspecific and variable symptomatology, hypoxic hepatitis is more commonly diagnosed incidentally with routine liver function tests anywhere from 2-24 hours after an episode of systemic hypotension. Laboratory abnormalities in hypoxic hepatitis are consistent with a hepatocellular pattern. First, there is a marked increase in aminotransferases and Lactate dehydrogenase (LDH), with AST and LDH rising most sharply and peaking in the first 12-48 hrs, while the rise and peak ALT is not as dramatic.50,59 Moreover, maximal elevation of ALT is less than AST and given the longer half-life of ALT, it reaches normal levels later than AST. Even so, the aminotransferase levels characteristically fall by greater than 50% within 72 hours of resolution of the underlying insult and return to normal within 7-10 days.34,50,60 Increases in LDH level tend to be massive and ALT/LDH ratio of less than 1.5 often distinguishes ischemic injury from other forms of acute hepatitis.61 It is of interest to note that the liver may not retain its normal architecture after regeneration if the reticular framework is damaged. The most common compensatory responses seem to be either thickening of hepatocellular plates with preservation of trabecular and cord-like pattern or nodular masses of hepatocytes.19,62 The latter, known as nodular regenerative hyperplasia, is less common but can manifest as a grossly granular or nodular liver.18
The integrity of hepatic synthetic function is also compromised in hypoxic hepatitis, as determined by PT/INR. If the INR remains above 1.5 despite adequate stores of Vitamin K, the diagnosis of acute liver failure is appropriate.63 Synthetic function is also of prognostic importance, with INR above 2.0 associated with an independent increase in mortality.50
Elevated lactate is also a common biochemical abnormality in hypoxic hepatitis although Fuhrmann et al. noted its lack of independent predictive value in mortality.50 Rarely, laboratory abnormalities can even include consumptive coagulopathy, which can be asymptomatic or symptomatic and most often related to the underlying etiology. Bilirubin may be mildly elevated and tends to peak after the transaminases and LDH levels begin to decline. The effects of systemic hypotension are not isolated to the liver, and increases in creatinine level from acute tubular necrosis are nearly universal early in the clinical course.
The differential diagnosis for hypoxic hepatitis includes acute viral hepatitis, autoimmune hepatitis, drug-induced liver injury (e.g. acetaminophen), acute Wilson’s disease and acute vascular thrombosis e.g. hepatic artery and portal vein thrombosis. While viral hepatitis and alcoholic hepatitis can usually be differentiated from hypoxic hepatitis based on the ALT:AST and AST:ALT ratios, respectively, it may be difficult to differentiate drug-induced hepatitis from hypoxic hepatitis.
Treatment
Early recognition and management is critical and is the primary prognostic factor. The importance of recognizing hypoxic hepatitis is underscored by reports of associated mortality in Intensive Care Unit (ICU) patients of over 50%.50,53 However, its role as an independent risk factor in ICU patients is still uncertain, as one report found that hypoxic hepatitis was only an independent risk factor in those requiring vasopressor therapy.50 Importantly, the cause of death is usually not due to hepatic failure but related to the underlying precipitating factor itself, such as sepsis or cardiac decompensation.50,53 Moreover, although encephalopathy is frequently noted in hypoxic hepatitis, it most often is not a true hepatic encephalopathy and is actually a consequence of the inciting factor leading to hypoxic brain damage.
Because this entity is essentially an observed laboratory abnormality, albeit an alarming one, treatment is targeted at identifying and addressing the inciting event. Awareness of potential exacerbating factors, such as mechanical ventilation or vasoconstrictors that may compromise hepatic blood flow, as well as metabolic monitoring to prevent derangements like hypoglycemia and lactic acidosis50 are essential. Also important is recognizing that other organs may be implicated, including initiation of systemic inflammatory response syndrome with possible disseminated intravascular coagulation,64 new or worsened respiratory compromise with hepatopulmonary syndrome,65 cardiac compromise with myocardial infarction, or renal compromise with acute kidney injury.
Several experimental therapies have been described. To improve hepatosplanchnic blood flow, infusion of renal-dose dopamine66 has been suggested, but to date no proven clinical benefit has been shown. Adenosine infusion has been used in animal models but there are no human data to support its use in this setting. Other investigators have suggested a role of antioxidants6 or N-acetylcysteine,67 however these findings are only limited to case reports and thus need to be corroborated in randomized controlled trials. Nitric oxide has shown some promise, given its role as an endothelin antagonist and consequent ability to counter vasoconstriction of hepatic vascular beds in ischemia.68 Similarly, research has been focused on angiotensin receptor II blockers and ACE inhibitors as possible antagonists of Renin-angiotensin-aldosterone system (RAAS) activation, a pathway very much implicated in hypoxic hepatitis.69 Molecular Adsorbent Recirculating System (MARS) and single-pass albumin dialysis, both of which have shown benefit in acute and acute-on-chronic liver failure, have also been researched as potential therapeutic modalities in hypoxic hepatitis but with uncertain benefit.70,71,72 To date, no liver-specific treatments have been proven to improve outcome. Furthermore, hypoxic hepatitis is not an indication for liver transplantation as the hepatic derangements are reversible with correction of the underlying disorder.
Prognosis
The majority of patients with hypoxic hepatitis follow a benign self-limited course with complete resolution of transaminases to normal values within 3 to 7 days of the inciting event.57 However, because this hepatic ailment occurs in the critically ill patients, survival in most series is rather poor. In the largest published series to date (142 episodes in 10 years of surveillance): the 1-month and 1-year survivals were 53% and 28% respectively.53 Fulminant hepatic failure rarely occurs and seems to be restricted to patients with longstanding congestive heart failure, cardiac cirrhosis45 or other forms of chronic liver disease.
CONCLUSION
Hepatic injury as a consequence of cardiac disease is a relatively common, but often poorly recognized, syndrome. An understanding of the hepatic circulation and normal liver architecture is important to appreciate how the hemodynamic changes of heart failure affects the liver, leading to the associated clinical, biochemical and histologic features. The hepatic manifestations of “congestive hepatopathy” and “hypoxic hepatitis” may range from mild liver enzyme abnormalities to progressive liver injury and, rarely, liver failure. A team approach characterized by collaboration amongst both cardiologists and hepatologists is critical for optimizing patient care and maximizing positive outcomes.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
PERSONAL ACKNOWLEDGEMENTS
The authors wish to thank Suganthi Soundararajan, MD, Department of Pathology, Drexel University College of Medicine, Philadelphia, PA for kindly providing the pictures.


