DIABETIC KETOACIDOSIS
Diabetic ketoacidosis (DKA) is an acute, major, life-threatening complication of diabetes which occurs in patients with either type 1 or type 2 diabetes.1 This condition is a complex disordered metabolic state characterized by hyperglycemia, ketoacidosis, and ketonuria. Insulin deficiency leads to the release of free fatty acids from adipose tissue, hepatic fatty acid oxidation, and the formation of ketone bodies, such as acetoacetate, 3-β-hydroxybutyrate (β-HB) and acetone (Figure 1). The observed pro-inflammatory and pro-coagulant states in hyperglycemic crises and hypoglycemia may be the result of adaptive responses to acute stress, and not hyperglycaemia or hypoglycaemia per se. 1
Usually, ketones are dealt with a risk factor of worse prognosis by ADA and other diabetes supporting organization.
FASTING AND HYPERKETONEMIA
In the fasted state, glycolysis is diminished; the flow of substrates entering the citric acid cycle drops, and ketone production are turned on. Cahill2,3 studied the glucose metabolism of people who let themselves fast for 40 days. He reported that in the starving human adult, β-HB and aceto-acetate are produced in the liver from long-chain fatty acids and β-HB could be the energy source in the brain and other tissues. A rise of β-HB blood concentration to approximately 6 mM was characteristic. The estimated glucose production at 5-6 wk of starvation was reduced to approximately 86 g/24 hr. Of this amount the liver contributes about one-half and the kidney the remainder. Approximately all of the lactate, pyruvate, glycerol, and amino acid carbons which are removed by the liver and kidney are converted into glucose, as evidenced by substrate balances across these organs.
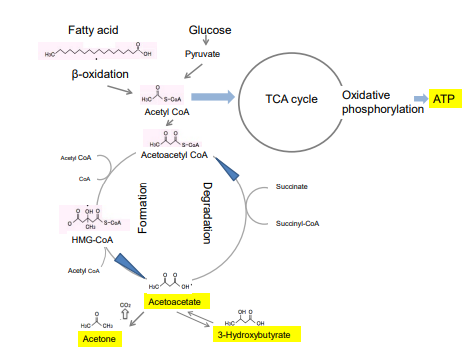
Cahill’s research was integrative rather than reductionism, but he opened the unique insight on the metabolic adaptation of humans to starvation. During starvation, extremely low insulin levels facilitate acyl-CoA entry into mitochondria, producing excess amounts of acetyl-CoA that cannot be metabolized in the Krebs cycle and are diverted towards the synthesis of ketone bodies (Figure 1).4 In 1960s, it was widely held that the brain did not oxidize ketone bodies for the production of energy. Cahill was one of the few clinical investigators at that time who believed that during starvation there was not enough nitrogen in the urine to account for the alleged amount of glucose that the brain was thought to need for normal function.5 Glucose, β-HB, and acetoacetate appeared to be used for energy sources of the brain of these people, as only ketone bodies increased without symptoms of ketoacidosis.5
Figure 1: Production of ketones and energy production. Ketone bodies, namely acetoacetate, acetone, and 3-hydroxybutyrate (βHB) are produced during an extensive fatty acid oxidation. They are the products of acetoacetyl-CoA and acetyl-CoA condensation. Rate limiting step of ketogenesis is the formation of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) and is catalyzed by mitochondrial HMG-CoA syntetase (HMGCS2). Utilization of ketone bodies as a source of energy, 3-oxoacetyl-CoA thiolase, and betaHD dehydrogenase are necessary. For degradation, succinyl-Co-A: acetoacetate-CoA transferase (SCOP) , also known as 3-oxoacid-CoA transferase is necessary. All of these enzymes are present in peripheral tissues at various levels, but SCOT is absent in the liver.
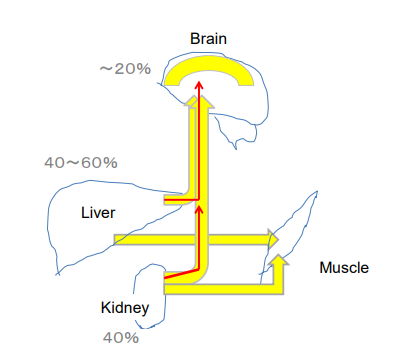
The tissues that produce β-HB include the liver, kidney and the brain astrocytes.6 As the brain consumes about 20% of its energy from glucose, similar amounts of ketones should be substituted to glucose to keep the brain functioning in the fasting state (Figure 2).
Figure 2: Glucose and ketone formation at fast or severe carbohydrate restriction. Hepatocytes, renal tubular cells and intestinal epithelial cells and astrocytes are able to carry out ketogenesis in prolonged fasting and ketogenic diets. After glucose is provided by gluconeogenesis in the liver and kidney (red arrow), ketone formation substituted (yellow). Astrocyte is considered to produce ketones up to 20% to support brain function.
Koda’s fasting and dietary therapy has proven effective for many intractable diseases in Japan.7 In an extreme case the blood concentration of β-HB remained above 3 mM without any clinical and laboratory symptoms.8 So, β-HB can substitute glucose as an energy source. It seems to stimulate the human nervous and endocrine systems, and to increase self-healing ability.
GESTATIONAL DIABETES
Gestational diabetes is not a rare complication of pregnancy. During pregnancy, the placenta produces high levels of various hormones. Almost all of them impair the action of insulin in maternal cells, raising the blood sugar level. Controlling the blood glucose can prevent birth complications and keep the baby healthy, so insulin therapy is usually tried in gestational diabetes.9
As the fetus grows, the placenta produces more and more insulin-blocking hormones. In gestational diabetes, placental hormones induce a rise in blood glucose up to a level that can affect the growth and welfare of the baby. Gestational diabetes is particularly severe in obese pregnant women. In such cases, insulin therapy is often ineffective, and doctors recommend abortion if elevated blood glucose levels fail to be under control.
American Diabetes Association (ADA) does not recommend a very low-carbohydrate diet where the uptake of carbohydrate is lower than 130 g per day. Muneta et al9 recently reported the cases of 16 gestational diabetic patients who had normal deliveries after a very low-carbohydrate diet (less than 5 g per diet). The blood level of ketone bodies and free fatty acids increased consistently, with a respiratory quotient (CO2 eliminated/O2 consumed) of 0.72, which means main energy comes from ketone bodies under eucaloric condition.10
Most patients were obese and lost body weight with the MEC ketogenic diet (100 g each of meat, eggs, cheese and leaves of green-yellow vegetables), blood glucose levels returned within normal range within a few weeks, and all deliveries were under control.
Maternal starvation in late gestation lowers insulin, and lipolysis supervenes. The continued glucose drain by the conceptus aids in converting the maternal liver to the ketogenic organ, and ketone bodies produced from incoming fatty acids cross the placenta to be utilized by the fetus. Muneta11 found high β-HB level (1 to 8 mM) among mothers, or in the umbilical cord blood, and also in the placental tissue fluid (Table 1).
Table 1: Concentration of β-hydroxybutyrate of placenta and new born peripheral blood.
|
Material
|
β-hydroxybutyrate |
Average |
Sample number
|
|
Chorionic Villi
|
600~4500 uM |
1930.1 |
98 |
|
Aborted Villi
|
600~3600 uM |
163.2 |
37
|
|
Umbilical cord
|
300~2500 uM |
779.2 |
60
|
|
Placental tissue
|
1200~5200 uM |
2235 |
60
|
| Peripheral blood on 4th day |
100~800 uM |
426.5 |
99
|
|
Peripheral blood at 1 month
|
200~700 uM |
366.7 |
24
|
High β-HB levels in the placenta have occasionally been reported by veterinarians, but most of them deals with domestic animals, where ketonemia is classically a sign of problematic delivery.12
DUAL ENGINE FOR ENERGY PRODUCTION
The β-HB and acetoacetate (AcAc) support mammalian survival during states of energy deficit by serving as alternative sources of ATP.2,3,4 Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase and HMGCS2 is the rate limiting enzyme of the ketogenic pathway (Figure 1).13,14,15 Succinyl CoA acetoacetateCoA transferase (SCDT) is necessary to convert β-HB to acetoacetate in the peripheral tissue cells. SCDT is lacking in hepatic cells, so, the liver is a major ketogenic organ but cannot use it. Animal species comparisons and biochemical data show that all fetuses can develop by using ketogenic energy through a pathway which seems to have been maintained throughout evolution.16 We thus hypothesized that β-HB could be a fuel for the basic engine that produces energy in all terrestrial species. However, ATP production from glucose-pyruvic acid pathway seems to become a dominant system in human. Why glucose has become the major fuel in the body? We hypothesize the establishment of TCA cycle in mitochondria and enough oxygen supply since 2 billion years ago would be the key reason. The efficacy of ATP production from β-oxidation product is 10 ATP molecules, while it is 12.5 molecules from pyruvic acid.17 So, more efficient burning system has developed by using various glucose transporters in various organs throughout evolution. We may call it a booster engine for energy production. Endosymbiosis with mitochondria, which should support the burning system with oxygen, is a miracle signs of life.
The liver and kidney contain abundant glycogen particles. These organs are also ketone producing organs, so it may represent a close relationship between ketone and glucose burning system (Figure 2). This explains why certain level of glucose is steadily maintained in the blood even in the hyperketonemic person. Some people could not raise ketone levels in the blood concentration as expected even by the ketogenic diet. A metabolic homeostasis would be different in these people, and needs further study.
THERAPEUTIC USE OF KETONE DIET
Epilepsy, Autistic Behavior and Childhood Obesity
Fasting and ketogenic diet was first introduced for the treatment of epilepsy by HR Geyelin and RM Wilder’s group in 1920’s.18 Over 250 medical centers worldwide offer ketogenic diets to children with epilepsy.19 However, access to these therapies has remained extremely limited for adults until recent years.20 From observations in 229 adults (age range of 18-86 years) attending the Adult Epilepsy Diet Center, ketogenic diet therapies appeared effective and safe in the long-term in adults.
The potential benefits of ketogenic diets are considerable. However, the effects of carbohydrate-depleted (ketogenic) diets on the metabolic parameters of children have been insufficiently assessed.21
To compare the efficacy and metabolic impact of ketogenic and hypocaloric diets in obese children and adolescents, fifty-eight obese subjects were assigned to one of two diets for 6 months. In both group participants significantly reduced their weight, fat mass, waist circumference, fasting insulin, and HOMA-IR, but the differences were greater in the ketogenic group. Only the ketogenic group had increasing in high molecular weight adiponectin.
Ketogenic diets may be used as a additional or alternative therapy in autistic behavior.22
β-HB and Prevention of Diabetic Complication
Myocardial infarction:
Cardiovascular events are common in diabetic patients. After infarct by the thrombosis, resuscitated blood flow by thrombolysis often damages remained tissue. Short-term fasting reduces the extent of myocardial infarction and incidence of reperfusion arrhythmias in rats.23 In experiments on cardiac ischemic tolerance in rats, short-term fasting increased the concentration of β-HB compared to controls. In addition, fasting limited the infarct size (48.5% of the area at risk) compared to 74.3% controls; the total number of premature ventricular complexes (12.5) was reduced compared to 194.9 controls; and the duration of ventricular tachycardia (0.6 s vs. 18.8 s) occurring at early reperfusion.
To investigate the role of high concentrations of β-HB in preventing heart damage after prolonged fasting, infarct size and the incidence of apoptosis caused by ischemia-reperfusion were determined in the Wistar rats.24 Apoptosis in the subendocardial region was significantly reduced in fasting. In addition, the levels of ATP in the fasting DL-β-HB treated group were significantly higher compared with control groups after 30 min of ischemia and 120 min of reperfusion.
Alzheimer and other nervous disease:
The Hisayama study25 started in 1961 is a prospective cohort study characterized by all dead people being autopsied. The prevalence of allcauses, dementia and Alzheimer’s disease (AD) significantly increased over time. Diabetes-related factors, such as fasting glucose, 2-hours post-load plasma glucose, fasting insulin, and homeostasis model assessment of insulin resistance (HOMA-IR) were measured in 1988.26 The results suggest that hyperinsulinemia and hyperglycemia caused by insulin resistance accelerate Alzheimer’s disease in combination with the effects of APOE epsilon.
Excess weight, especially abdominal obesity, can cause or exacerbate cardiovascular and metabolic diseases. Obesity is also a proven risk factor for AD.
Various studies have demonstrated the beneficial effects of a ketogenic diet in weight reduction and in modifying the disease activity in neurodegenerative disorders, including AD.3,27-30 Compared with obese rats fed a control diet, obese rats fed a ketone diet showed significant weight loss, improvements in lipid profiles and insulin resistance, and up-regulation of adiponectin mRNA expression in adipose tissues. In addition, the ketone diet triggered significant down-regulation of the expression of brain amyloid protein precursor, apolipoprotein E and caspase-3n, and improved brain oxidative stress responses. These findings suggest that a ketone diet has anti-obesity and neuro-protective effects.
Caprylic acid triglyceride is registered as a therapeutic food supplement apparently effective for the treatment of Alzheimer’s disease in the United States.31 Caprylic acid is a middle chain fatty acid (molecular formula C8 H16O2 with eight carbon atoms). The middle chain fatty acid can penetrate blood brain barrier, so it becomes substrate of beta oxidation, which increases the production of the derived ketone body in astrocytes.32
Evidence suggests that energy production in the nervous system increases in parallel with symptomatic improvement. In addition, the protection of nerve cells involves adjustments in gene expression, as well as multi-inflammatory, anti-oxidation, and anti-apoptosis mechanisms (later description). β-HB also improves cognitive functions by increasing brain perfusion.
Aging is associated with an increased susceptibility to hypoxic or ischemic damages and a decline in behavioral functions which may be due to attenuated adaptive and/or defense responses.28 Diet-induced ketosis could improve the behavioral performance of aged rats. For example, old Fischer rats were fed either standard or ketogenic (KG) diets for 3 weeks, and then exposed to hypobaric hypoxia. Cognitive function was measured using the T-maze and object recognition tests. KG diet significantly increased blood ketone levels in both young and old rats. In the aged rats, the KG diet improved cognitive performance under normoxic and hypoxic conditions. Capillary density and HIF-1α levels were elevated in the aged ketonic group, independently of hypoxic challenge. These data suggest that diet-induced ketosis may be beneficial in the treatment of neurodegenerative conditions.
Pharmaceutical Mechanisms of β-HB Action
Inhibition of histone deacetylase:
Shimizu et al33 report that the ketone body β-HB is an endogenous and specific inhibitor of class I histone deacetylases (HDACs). Histone acetylation is a prominent epigenetic modification of the central nervous system that is unequivocally associated with an increase in the rate of gene transcription. Histone acetylation generally favors long-term memory. Histone acetylation is also amenable to pharmacological interventions, predominantly the use of histone deacetylase (HDAC) inhibitors (Figure 3)34-37 It has therefore spurred considerable interest as a putative target of cognitive enhancement.
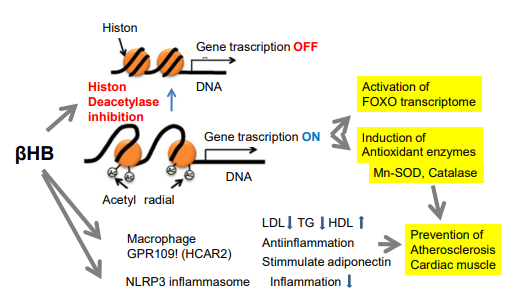
Figure 3: Epigenetic activity as inhibitor of histone deacetylases leads to various effects. Activity as an inhibitor of histone deacetylases leads to various epigenetic modulations, associated with global histone hyperacetylation. Induction of stress response gene FOXO leads to reactive oxygen species detoxification, apoptosis, etc.
Because of the ubiquitous presence of histone acetylation, HDAC inhibitors have great potential not only to treat cognitive impairment resulting from neuro-degenerative disorders, but also to serve as cognitive enhancers for the healthy ones. Gräff and Zsai34 reviewed the state of the art of HDAC inhibitors used as cognitive treatments or cognitive enhancers. They describe epigenetic priming as a new model for their mode of action, caution against their unsupervised usage, despite their overall great promise.
Recent evidence indicates that the inhibition of histone deacetylase (HDAC) protects the heart against myocardial injury and stimulates endogenous angiomyogenesis, even in the diabetic heart.35 Sodium butyrate (1%), a specific HDAC inhibitor, was added daily to the drinking water in streptozocin induced diabetic mice to inhibit HDAC activity. HDAC inhibition resulted in a significant functional improvement in STZ-injected diabetic mice. Likewise, HDAC inhibition attenuates cardiac hypertrophy, as evidenced by reduction of heart/tibia ratio and in areas of cardiomyocyte distribution. This was associated with reduced interstitial fibrosis, a decrease in caspase-3 activity and apoptotic histochemical staining, but also with increased angiogenesis in diabetic myocardium. Notably, glucose transporters (GLUT) 1 and 4 were up-regulated following HDAC inhibition, which was accompanied with increases of GLUT1 acetylation and p38 phosphorylation. Furthermore, myocardial superoxide dismutase, an important antioxidant, was elevated following HDAC inhibition in the diabetic mice.
Class I HDAC inhibition elicits the protection of contractile function following ischemia reperfusion. The study highlights the need for the development of new strategies that target specific HDAC is forms in cardiac insufficiency.36
In the heart, the enhancement of lysine acetylation or SUMOylation using HDAC inhibitors or SUMO-1 gene transfer respectively, has been shown to be cardio-protective.37 The treatment of cardiomyocytes and cardiac fibroblasts with pharmacological inhibitors of HDAC catalytic activity robustly increased the conjugation of SUMO-1 with several high molecular weight proteins in both cardiac cell types. The use of a battery of selective HDAC inhibitors and short hairpin RNAs demonstrated that HDAC2 is the primary HDAC isoform that controls cardiac protein SUMOylation.
Stimulation of FOXO:
The fast controls growth hormones, insulin and IGF-1 serves as signals in the transduction system and causes the acetylation of histones in the promoter domain of the FOXO3 gene, inducing the expression of FOXO3.38 It induces resistance to stress by raising the transcription activity of FOXO.
A transcription factor called FOXO3 is activated at the time of starvation. FOXO3 is able to raise the resistance to oxidation stress and starvation stress. FOXO is a transcription factor belonging to the subgroup “O” of the Forkhead family, interacting DNA-binding domain FOX (Forkhead box) by abbreviation of “Forkhead box O”.
In addition, β-HB acts in GPR109A developing in a fat cell and macrophage, which leads to improve arteriosclerosis (Figure 4). The action increases the expression of antioxidant enzymes, such as SOD or catalase, protecting myocardium against oxidation injury.
Resistance to oxidative stress:
Concentrations of acetylcoenzyme A and nicotinamide adenine dinucleotide (NAD (+)) affect histone acetylation and thereby couple cellular metabolic status and transcriptional regulation. The administration of exogenous β-HB, or fasting or calorie restriction, two conditions associated with increased β-HB abundance, all increased global histone acetylation in mouse tissues.30,33 The inhibition of HDAC by β-HB was correlated with pleiotropic changes in transcription, including transcription of genes encoding oxidative stress resistance factors FOXO3A and MT2. The treatment of cells with β-HB increased histone acetylation at the FOXO3A and Mt2 promoters, and both genes were activated by selective depletion of HDAC1 and HDAC2. Consistent with increased FOXO3A and MT2 activity, the treatment of mice with β-HB conferred substantial protection against oxidative stress.
Protection of mitochondria:
It is known that the improvement of the oxidative phosphorylation within mitochondria is important for the cell protection of nerve cells from injury and the reinforcement of cognitive functions. In neuronal energy metabolism, mitochondrial function is important for the treatment of dementia, such as Alzheimer’s disease and other neurodegenerative diseases.
Zang et al39 reported that the mechanism whereby β-HB methyl ester (HBME), becoming β-HB of the ketone body by ingestion in the body, protects mitochondria.
Alzheimer’s disease (AD) is caused by multiple mechanisms, including a decrease in the cellular utilization of glucose, and mitochondrial alterations in brain cells. HBME inhibits cell apoptosis under conditions of glucose deprivation, and it rescues the activities of mitochondrial respiratory chain complexes that are impaired in AD patients. HBME stabilizes the mitochondrial membrane potential. AD mice treated with HBME demonstrated that HBME has a positive in vivo pharmaceutical effect to improve the spatial learning and working memory of mice. A reduction in amyloid-β deposition in mouse brains after intra-gastric administration of HBME was also observed.
When the concentration of blood β-HB was raised between 0.6-1.5 mM in the mice by giving fast and direct β-HB, it was confirmed that the acetylation of histones increases in multiple organs, including the kidney.
Stimulation of adiponectin release:
Niacin (nicotinic acid) has recently been shown to increase serum adiponectin concentrations in men with the metabolic syndrome. Since niacin appears to exert its effects on lipolysis through receptor (GPR109A)-dependent and -independent pathways, the role of the identified GPR109A receptor in adiponectin secretion is noteworthy.40 As niacin administration had no effect on adiponectin and NEFA concentrations in the GPR109A receptor knockout mice, the GPR109A receptor plays an important role in the dual regulation of adiponectin secretion and lipolysis. β-HB has the similar effects on GPR109A receptor.
Suppression of inflammasome:
It becomes clear that the inflammasome plays an important role in the onset and the progress of type 2 diabetes, Alzheimer’s disease, arteriosclerosis and inflammatory diseases, in addition to a number of autoimmune diseases. The inflammasome activates inflammatory caspase and cytokines of the IL-1 family within a complex of proteins involved in inflammation and apoptosis.38 The inflammasome has been seen as a natural immunity system that protects living organisms against alien substances and pathogenic microorganisms.
Prolonged fasting reduces inflammation. However, we do not know what effect on the innate immune response result from ketones, and other alternative metabolic fuels produced during energy deficits. β-HB, contrary to AcAc and the structurally related butyrate and acetate, suppresses the activation of the NLRP3 inflammasome in response to urate crystals, ATP and lipotoxic fatty acids. Mechanistically, β-HB inhibits the NLRP3 inflammasome by preventing K (+) efflux and by reducing ASC oligomerization. The inhibitory effects of β-HB on NLRP3 are not dependent on chirality or starvation-regulated mechanisms like AMP-activated protein kinase (AMPK), reactive oxygen species (ROS), autophagy or glycolytic inhibition. β-HB reduces NLRP3 inflammasome-mediated interleukin IL-1β and IL-18 production in human monocytes. The anti-inflammatory effects of caloric restriction or ketogenic diets may be linked to β-HBmediated inhibition of the NLRP3 inflammasome.41
FUTURE PROBLEM
A new therapeutic approach could merge for the treatment of cancer.42-47 Metformin is usually used for the treatment of type 2 diabetes. Recently, metformin, vitamin D and ketone in combination showed broad-spectrum antitumor activities.48 In combination, metformin and vitamin D exhibited synergistic effects on cancer cell proliferation and apoptosis. The underlying anti-tumor mechanisms may involve m-TOR related pathways, which are related to activating expression of cleaved caspase-3, Bax and p-AMPK.
However, many problems remain. Humans have no experience to delete carbohydrate from meal at all, and high protein and lipid intake should be inevitable to compensate total energy expenditure. The balance of these effects should be studied, but notably original ketogenic diet for epilepsy by Russel Wilder in 1920 contained nearly 90% fat. High protein diet is a risk factor of cardiovascular diseases, renal insufficiency and cancer.49 So, the balance of risk and benefits should be considered.50
Finally, the interaction with intestinal microbiota should be clarified. We have previously hypothesized that bifidobacterium contribute to produce β-HB.5 There are many bacteria that can synthesize poly(3-hydroxy butyrate-co-3 hydroxyvalerate) oly-beta hydroxyl butyrate.51 These bacteria are capable of using a broad range of carbon sources for their growth and for the production of polyhydroxyalkanoates (PHAs). They can use monosaccharides (glucose and fructose), disaccharides (sucrose), pentoses (xylose and arabinose), various organic acids (acetic acid, propionic acid and octanoic acid) and even the acid pre-treated liquor (APL) of sugarcane trash, a lignocellulosic biomass, for growth and the production of polyhydroxyalkanoates (PHAs).
The contribution of intestinal microbiota to ketone body production should open a new field of medicine.
ACKNOWLEDGEMENT
The authors appreciate Dr. Philippe Culain, MSF, for his kind English revision and discussion.
CONFLICTS OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflicts of interest.


