INTRODUCTION
The outgrowth of a clonal population of cells that follows a loss of tissue homeostasis is one of the hallmarks of cancer development (Figure 1). Tissue homeostasis/balance between cell birth and death is regulated by a several processes such as cell cycle arrest and apoptosis. Defects in these pathways clearly cause a selective growth advantage. Several cancers possess inactivating mutations in the genes that involve the checkpoints that keep the cell cycle events in their appropriate order1 and influence apoptotic pathways,2 meanwhile some cancer-derived cell lines show a resistance to apoptotic stimuli. Recently, experimental evidence supports the suggestion that genetic modifications that inactivate apoptotic pathways are actively selected during tumour growth.3
Cell death activated by anticancer therapeutic agents might be due to delayed events which are genetically determined and still different from the more direct apoptotic program. A considerable number of indirect proofs support the hypothesis that the initiation of the process of apoptosis in cancerous cells is significant to their successful removal by therapeutic agents.2 Increasing understanding of the whole process of apoptosis has been due to the advancement in the discovery of vital molecules involved in the death pathway. Intense research has showed the occurrence of many interrelated but different pathways that lead to apoptosis (programmed cell death). Notably, a cell type defective in all of these pathways has not been identified.
Figure 1. Hallmarks of Anastasis
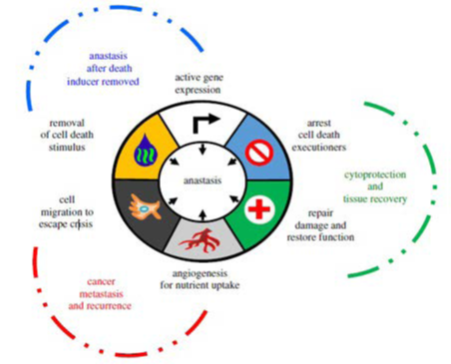
Adapted from Bell et al6
Apoptosis is an established form of regulated/programmed cell death, with different signalling pathway; however, the effect or molecules or the eventual executors of apoptosis are the caspases. The caspases cleave the structural proteins eventually leading to cell death after the caspases has been activated. During apoptosis, gross segmentation of essential cellular organelles was noted, this gives the general insight that the apoptotic pathways are irreversible.4 Contrary to this, recent studies show that cells committed to apoptosis can be stabilized and can be reverted (anastasis).4,5
In this review, we collate and summarize evidences supporting anastasis, with probable mechanisms of action. Given the newness of this focus, we look forward to possible physiological, pathological and therapeutic implications of anastasis, and suggest future directions for the study of anastasis.
Apoptosis: Programmes Cell Death
The term programmed cell death (PCD) was introduced in 1964, and can be referred to as cell death during development; it is not an accidental process, but rather follows numerous controlled steps resulting to a defined self-destruction (Figure 2).
Figure 2. Overview of Extrinsic and Intrinsic Pathways of Apoptosis
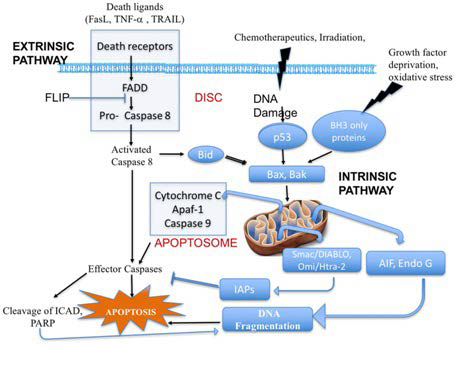
Adapted from Marin et al20
Cancer known as a complex genetic disorder resulting from mutations of oncogenes or tumour suppressor genes, leading to the alteration of vital signalling pathways, has been connected to PCD. Interpreting programmed cell death in disease conditions is necessary because it does not only show novel insights into the pathogenesis of such conditions, but also help in developing new targeted anticancer therapeutic approaches.
Inequity among cell division and cell death is one of the major deregulated landmarks in cancer. Also, other factors that affect cell survival, proliferation and differentiation are essential in cancer. From this point of view, any modifications in cell homeostasis or development may lead to dysregulation, with PCD to decide its fate, since it can hold the equilibrium between cell death and survival, based on the trophic conditions of the cell.7 Though, this is a double-edged sword as PCD can be the cause of and the solution to the problem.8 Thus, programmed cell death plays a vital role in both the development and treatment of cancer. The three major forms of programmed cell death that can be differentiated by their morphological and physiological differences are apoptosis, autophagy, and programmed necrosis/ necroptosis9 and they may together decide the fate of the cancer cell. Although, another types of cell death might be exploited in future to control and eliminate cancer cells, therefore showing clue towards a new therapeutic strategy.
Programmed cell death is an indispensable component of life. With over 20 forms of programmed cell death that have been suggested,10,11,12 apoptosis is the most well-known and studied for its regulatory mechanisms in cell suicide, and important roles in embryogenesis and normal homeostasis by removing the useless, injured or dangerous cells in the body.13 Meanwhile genetic or pharmaceutical strategy allow dying cells to survive that otherwise would normally die14,15 induction of apoptosis is generally characterized by an irreversible commitment to cell death.16 Events canonically marking this irreversible or point of no return in apoptosis include mitochondrial cytochrome c release into the cytosol (Figure 2),17 activation of execution caspases,18,19 and their related or downstream events, like cell shrinkage, deoxyribonucleic acid (DNA) damage, cell body fragmentation, cell surface exposure of ‘eat me’ signal. Meanwhile, an increasing number of evidences confirmed that dying cells can exit the initiated death process and recover, even at late stages known as beyond the ‘point of no return’.
THERAPEUTIC POTENTIAL ANASTASIS
The word anastasis, which means ‘rising to life’ in Greek,19 to depict the recovery of dying cells after verge of death, using reversal of apoptosis as the first example. Recently, studies showed that dying cells can recuperate after important cell death hallmarks of apoptosis and possibly other mode of cell death process. Elimination of the death stimulus is adequate to allow anastasis to take place both in vivo and in vitro, demonstrating that anastasis is an intrinsic recovery event (Figure 3).
Figure 3. Recovery from the Brink of Death
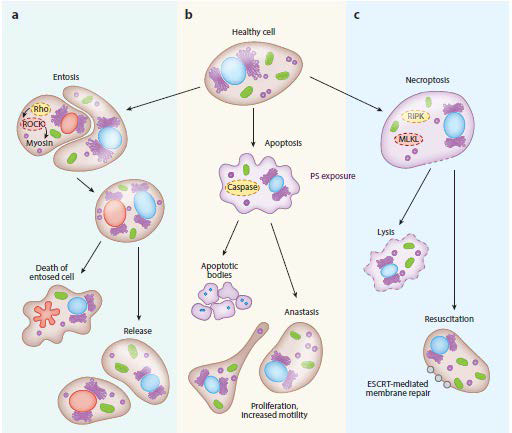
Adapted from Sun et al4
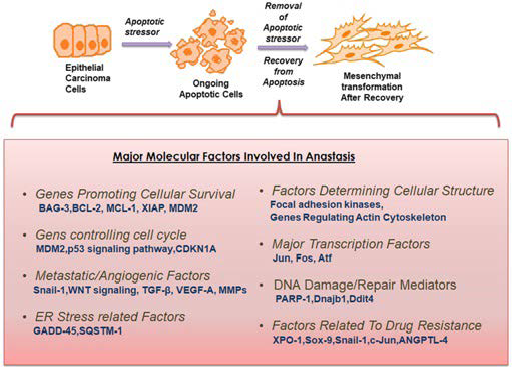
Anastasis pathway involves the removal of the apoptosis inducing agent, the cells recover to a stable state.19 Molecular analysis of anastasis illustrated comparison to wound healing in the form of upregulated receptor tyrosine kinase, converting growth factor-beta, mitogen-activated protein kinase (MAPK) signalling and angiogenesis-promoting pathways. Similar to any repair mechanism, a minor population of the recovering cells from apoptosis frequently hold some form of sub-cellular damage which could reduce the functionality of the cell and impart a genetic instability resulting to oncogenic transformation.21 To validate the possible effects of anastasis, researchers must decipher the genomic profile of normal cells recovering from apoptosis. In these cells, it is imperative to examine if the apoptosis has induced genetic abnormalities influencing carcinomatous conversion resulting to carcinogenesis.
During apoptosis, changes such as induction of cell shrinkage, membrane blebbing and formation of apoptotic bodies initiate concurrent accumulation of messenger ribonucleic acids (mRNAs) coding for anti-apoptotic and survival factors including the zinc-finger transcription factor Snail4 ; this survival coded mRNAs are being accumulated and are not translated on to the cells until the apoptotic inducing factors subside. These results present a clear suggestion that apoptosis is potentially reversible, even though at present it is impossible to envisage if the recovering cells would be stable or suffer genetic instability and undergo an oncogenic transformation. Also, the regular assumption was that these stored mRNAs will be released after apoptosis and will be used by the surrounding cells for their survival and growth. With respect to cancer, it was known that the surrounding cancer cells could use the released mRNA of the apoptosed cancer cells to promote their metabolic activity resulting to cancer progression. Contrarily, anastasis inhibits cell death, the recovering cells may utilize its own stored mRNA for recovery and in case of recovering cancer cells, it would serve as an extra energy source for increasing its proliferative, metabolic and invasive potential.
Anastasis-based recovery of apoptotic cells has been indicated to take place in two phases. First, the initial phase, the apoptotic cells which are growth arrested was reverted to a steady state with growth possibility. Second, the later stage, anastasis enhanced numerous tumorigenic properties of the recovered cells including epithelial mesenchymal transition (EMT), proliferation, angiogenesis, and migration.
Similar to Sun et al4 investigation on cancer cells, the molecular biology of anastasis in a normal cell should be observed to detect possible molecular aberrations that might prompt the cells to go through a carcinomatous transformation. With genetic aberrations, enhancing the apoptotic signals in target cells could overcome the cell’s anastasis potential.
With respect to cancer cells, the anastasis might be a survival method against apoptosis induced by therapeutic agents. Furthermore, during anastasis additional genetic aberration could induce more resistance to therapy in recovering cells. Therefore, recovering cells through the anastatic mechanism are at the risk of being more aggressive and resistant to therapeutic modalities.
Stem cells have a longer life span and innate capability to resist apoptosis, making them to have a larger potential for anastasis than other cells. Hence, in cancer, targeting the anastasis in cancer stem cells (CSCs) might increase its sensitivity to therapeutic modalities. Adequate evidence proving anastasis as a mechanism of cancer progression or initiation, then inhibiting anastasis directly or indirectly by upregulating the apoptotic signals could serve an efficient therapeutic scheme.
Effect of Activated Caspase-3
Activation of caspase-3 has been attributed to a point of no return in terms of activation of apoptosis, however, recent studies contradict it. A range of cultured mammalian cells, such as cell lines, primary cells and cancer cell may survive caspase-3 activation via anastasis.19,22 Chemically induced apoptotic cells show typical morphological signs for example blebbing and shrinkage, and also biochemical hallmarks including exposure to phosphatidylserine and caspase-3 activation. If the cells are left in the toxins/chemicals, the treated cells die. But, if the cell death inducer is removed, most of the cells treated recover normal morphology, survive, and divide.4
Identification of factors essential in anastasis could have important implications in disease. Chemotherapy and radiation as cancer therapy induce apoptosis but are delivered rapidly, due to their toxicity. Thus, if even a portion of tumour cells undergo anastasis in vivo during or after treatment, they could sow the seeds for degeneration. Survival from caspase activation can also result in oncogenic transformation and genetic instability, signifying that anastasis may contribute to the initiation of tumour.19,21,23,24
In contrast to the possible detrimental consequences of anastasis mechanism in cancer, in normal cells, particularly those that are difficult to replace for example cardiomyocytes or neurons, anastasis may help preserve cells after transient injury. In support of this suggestion, cardiomyocytes appear to undergo anastasis in vivo following transient ischemia.25 In addition, a mouse model of τ-mediated neurodegeneration, over-expression of a mutant form of human tau protein triggers caspase-3 activation in neurons. Concurrently live imaging of caspase-3 activity and tau tangles in the brains of mice reveals that cells with transient activity of caspase can survive long-term.26
In a probably related course, a limited population of mitochondria permeabilize in a process called minority mitochondrial outer membrane permeability (MOMP), sub-lethal caspase activation, promoting tumorigenesis and transformation.27 Although, minority MOMP will not results in adequate activation of caspase resulting in the morphological hallmarks of apoptosis. Hence, both anastasis and minority MOMP can result to survival of cells with DNA damage and oncogenic transformation (Figure 4).
Figure 4. Schematic Representation of the Structural and Molecular Changes during Anastasis
Adapted from Iwakuma et al28
Activated caspases bring out the proteolytic damage of main functional and structural components of the cell during apoptosis execution phase. This massive and rapid cellular destruction is initiated and enlarged via several apoptotic pathways mediated by caspases. Activation of caspase proteases eventually results to organelles fragmentation, exposure of phagocytic signals on the cell surface, destruction of the genome by activating apoptotic DNases27 and also formation of apoptotic bodies for phagocytosis of dead cells.29,30
Cleavage of BID by activated caspase proteases subsequently results in the translocation to mitochondria promoting MOMP in order to enhance the apoptotic cascade.
With the significant effect of caspases in initiating and enlarging death signals that destroy or cripple numerous cellular processes, apoptosis is known to be unavoidable after caspase activation.18 Fascinatingly, recent findings reveal that apoptosis reversal can actually take place after caspase activation in cultured cells and in live animals. Caspase activated cells can also recuperate after cytosolic and nuclear condensation, plasma membrane blebbing and cell shrinkage, fragmentation of the mitochondria,31 thus demonstrating that apoptosis can be reversible at the cell execution stage (Figure 4).
Technically, it is difficult to express anastasis in animals, because cells can be morphologically identical from nearby healthy cells following anastasis. An in-vivo Caspase Tracker biosensor which is activated by effector caspase to activate permanent fluorescent protein expression within anastatic cells and their progeny, allowing the tracking of cell fate was developed to overcome this challenge (Figure 5).3
Figure 5. Genes Up-regulation and Possible Related Pathways during Anastasis
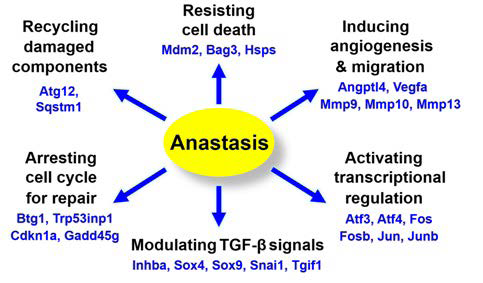
Adapted from Salameh et al32
Mouse double minute 2 (MDM2) of expression also activates the X-linked inhibitor of apoptosis protein (XIAP),33 arresting the death cascade by inhibiting the activated initiator and executioner caspases and enhancing the degradation of second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (Smac/DIABLO).34 Transcription is also very active during anastasis, generating building blocks for the recovering cells to repair damage and regain normal morphology. Upregulation of potent angiogenic factors, such as vascular endothelial growth factor A (VEGFA) and angiopoietin-like 4 (ANGPTL4), which promote angiogenesis and vascular permeability were also displayed by anastatic cells.35 This can promote anastasis by facilitating the nutrient supply and elimination of cellular wastes including those produced by degradation of the caspase-cleaved products.
During anastasis, up-regulation of the ubiquitin ligase MDM-2 takes place, resulting to degradation of the tumour suppressor gene p53, hence suppressing the pro-apoptotic p53- mediated DNA damage response. Heat shock protein (HSP)-70 upregulated during anastasis may also interact with and suppress apoptotic inducing factor (AIF) and Endonuclease (Endo)-G to stop DNA destruction initiated after MOMP. While being cleaved by caspases during apoptosis, the expression of inhibitor of caspaseactivated DNase (ICAD) and poly (ADP-ribose) polymerase (PARP) has been observed to reverse to pre-apoptosis stage during anastasis, allowing ICAD to hinder the DNase effect of CAD, and PARP to repair DNA damage in anastatic cells. Besides, cell cycle arrest is vital for DNA repair,36 and up-regulation of the cell cycle arrest genes including Cdkn1a, Trp53inp1 and Btg1 takes place during anastasis (Figure 6).
Figure 6. Possible Implication of Anastasis37
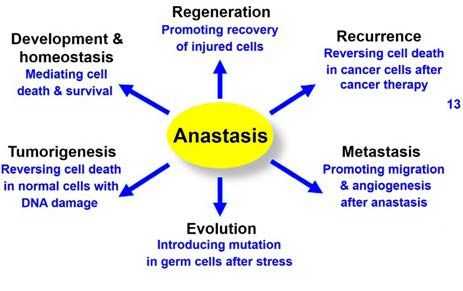
Adapted from Tang et al37
Reunion after Formation of Apoptotic Bodies
Interestingly, anastasis do occur even after the fragmentation of the dying cells.31 Recovered cells also reveal an increased number of micronuclei and chromosomal abnormalities, indicating unrepaired DNA damage after anastasis.36
Increased aneuploidy occurs when apoptotic bodies failed to truly reassociate with broken chromosomes during anastasis. It is uncertain how fragmented apoptotic cells can unite to an apparently normal structure. Recent findings have shown that surface exposure of phosphatidylserine may be the main factor for different types of cell fusion, such as formation of myotube and macrophage polykaryon, viral infection and fusion of severed axons.38 This results to assumption that the fragmented cells with externalization of phosphatidylserine could utilize this approach for repair and fusion during anastasis.
Under normal physiological conditions, apoptotic body is formed after phagocytosis. Internalized living cells undergoing entosis have been noted to escape their cannibalized state, eventually surviving and proliferating (Figure 3).39 Though entosis is not usual phagocytosis and is characterized by an invasion of a living cell into the cytoplasm of another cell, and so it may not represent how macrophages engulf apoptotic cell fragments.40,41
Effect of Pro-Survival and Pro-Metastatic Genes in Anastasis
Chemotherapeutic agents induce cellular stress leading to activation of pro-survival genes which eventually oppose entry into apoptosis. This intensive effort to bar apoptotic signalling has usually been thought to only be efficient before the apoptotic process is fully initiated.41 Recently, reports suggested that the mediators of cellular survival can revive cells from late stages of apoptosis, hence disregarding the notion of a “point of no return” from apoptosis, giving a chance to study the complexities of anastasis.42
Cancer cells survival does not only rely on the expression of anti-apoptotic genes, though they play a significant role, and also on genes responsible for DNA repair, cell cycle regulation, transforming growth factor beta (TGF-β) modulation, angiogenesis and migration. These factors are hyper-activated during early anastatic responses. The increase of survival factors, such as MDM-2 (Figure 5), during anastasis shows the likely involvement of p53 and its related signalling opponents including E2F transcription factor, cyclin-dependent kinases and cell cycle regulatory elements during the initial recovery period.43
Additional studies associated to activation of activator protein 1 (AP-1) transcription factors such as c-Fos and c-Jun are crucial, because these genes can either be pro-survival or proapoptotic based on the cellular context; thus, anastatic activation of these transcriptional factors give indications for the balance between pro-apoptotic and pro-survival mechanisms.44 Most of the survival proteins involved in anti-apoptosis and pro-proliferation also partake in the recovery from the verge of cell death. This implies that the capability of cancer cells to activate survival mechanisms, even in severe stress situations, leading to major molecular and structural changes to revert to its normal physiological state.
Structural adjustments during anastatic process include activation of focal adhesion kinases and rearrangement in the actin cytoskeleton.44 These structural changes are highly related to cellular migration; therefore, the cytoskeletal alterations in the late phases of anastasis aid cellular migration in recovered cancer cells. Cancer stem cells (CSC) are a matter of concern for the development of emerging therapies as they are highly related with tumour relapse and drug resistance. Likewise, anastatic cells represent a population of resilient cells defying different phases of apoptosis (Figure 6). More studies to evaluate the implication of anastatic mechanisms in genesis of cancer stem cells are required.
CONCLUSION
The relationship between anastasis and tissue recovery, progression of diseases and cell death decision remain to be elucidated. Additional work is required to establish whether anastasis is involved in these processes. If this is true, studies on the physiological and pathological roles of anastasis could give novel perception into multidisciplinary fields of research that helps our understanding in the control of cell death and survival, and also provide ability to recognize new therapeutic strategies for cancers, cardiopathies, degeneration, tissue injury and regeneration medicine by mediating reversibility of cell death processes.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.


