INTRODUCTION
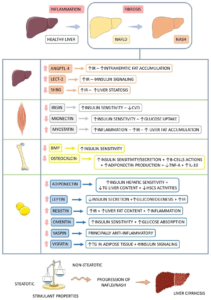
Non-alcoholic fatty acid liver disease (NAFLD), by definition, is macro-vesicular steatosis (fat accrual) is greater than 5% of the hepatocyte without any other etiology like alcohol or any prior liver disease. Non-alcoholic steatohepatitis (NASH) occurs as a complication of NAFLD. Besides steatosis, it possesses the properties of lobular inflammation along with the escalated diameter of hepatocytes (ballooning of hepatocytes) with or without fibrosis and implicates about 20% of subjects with NAFLD. This type of alteration has been noticeable on histological evaluation in addition to the presence of fibrosis, which displays the deterioration of the case. The propagation is correlated with cirrhosis, hepatocellular carcinoma (HCC), and liver transplantation, along with escalated mortality due to liver reasons.1 NAFLD is believedto be the hepatic constituent of metabolic syndrome (MetS), besides being intricately associated with metabolic diseases like obesity, type 2 diabetes mellitus (T2DM), and atherosclerosis.2,3 Based on these characteristics, an International Expert Consensus Statement pointed out that utilization of a new definition should be made regarding metabolic impairment associated fatty liver disease (MAFLD).4 The diagnostic criteria advocated were the existence of hepatic steatosis, obesity, and T2DM, or metabolic impairment.5 Despite this, it would express the intricate association of fatty liver with excessive feeding, sedentary lifestyle, and medical situations (like hypertension, T2DM, or dyslipidemia), and obesity.6 Nevertheless, no consensus was reached regarding these criteria, which were believed to be irrelevant regarding clinical scenarios since certain non-obese non-diabetic patients with hepatic steatosis were not diagnosed in view of the absence of laboratory investigations for metabolic impairment.6
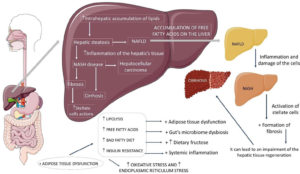
The insight regarding the pathophysiology of NASH was generated from the lucrative theory in the etiopathogenesis of NAFLD, namely the “2hit” posit.7 The first hit is insulin resistance (IR), secondary to escalated fatty acid (FA) flux; the second is inflammation, correlated with gut-obtained endotoxins, oxidative stress (OS), and mitochondrial impairment; however, it was not enough to detail the numerous molecular pathways along with metabolic events in NASH formation. Hence, a “multi-hit” model was posited more recently in view of it taking into account various damages taking place simultaneously in genetically prone subjects (Palatin-like PhosphoLipaseA2Domain-Containing 3 (PNPLA3), along with TM6SF2 genes) like nutritional, intestinal microbiota IR, hormones liberated by adipose tissue (AT), genetic, and epigenetic factors (Figure 1).1,8-11
Figure 1. Non-alcoholic Fatty Liver Disease and its Progressions
Adopted from Margiotti dos Santos et al11 increase, +: plus, NAFLD: non-alcoholic fatty liver disease, NASH: non-alcoholic steatohepatitis.
The determination of NASH prevalence is tough in view of the need for a liver biopsy, which is the gold standard for diagnosis, though this is not performed routinely. Indirect pointers state that 3-6% of Americans manifest NASH, with greater prevalence in subjects with prior-generated metabolic diseases, with 20% propagating to cirrhosis.1,12
Taking into account that NAFLD impacts as many as 1 billion individuals globally and that its prevalence is correlated with escalating obesity and MetS, it has been predicted that a set of both these disorders (NAFLD and NASH) would be implicated as the major etiology of liver transplantation.8,12
As far as the etiology is concerned, the primary factors correlated with NAFLD and NASH are: i) IR; ii) obesity; iii) T2DM; and iv) MetS. Other endocrine diseases like hypothyroidism and polycystic ovary syndrome PCOS) can be correlated with them in view of their sharing the elemental pathophysiological modes of NAFLD, IR, obstructive sleep apnea syndrome, hypertension, dyslipidemia, alterations in the intestinal microbiota, genetic proneness, menopause, a lifestyle with decreased physical activity, and an escalated intake of fructose and saturated fatty acids along with carbohydrates.1,13
In view of liver impairment, the liberation of hepatokines further alters. Furthermore, the correlated metabolic alterations result in changes in the liberation of other organokines that further modulate NASH in the etiopathogenesis and propagation of the disease.14 Earlier, we reviewed different methods of treating NAFLD and NASH and concentrated on organokines liberated by AT and the liver, which are key organs for controlling lipid metabolism, basically adipokines and hepatokines.15–32Furthermore, we reviewed the role of adipokines in obesity with heart failure (HF) and those of myokines like irisin in exercise regarding obesity15,16 and thought they might work in the form of diagnostic and therapeutic targets in NAFLD and NASH obtained HCC. These are believed to be biological markers that can anticipate the robustness of
NAFLD from NAFLD to HCC. In view of these, we decided to review more detailed parts of organokines in NASH propagation, inclusive of osteokines like bone morphogenetic protein (BMP), osteocalcin, and myokines in NASH and their crosstalk impacting the disease, besides overlap of certain organokines as both adipokines and myokines, or say adipokines and hepatokines, adipokines, and osteokines occur.
METHODS
We conducted a narrative review utilizing search engines PubMed, Google Scholar, Web of Science, Embase, and the Cochrane Library utilizing the MeSH terms “NAFLD”, “NASH”, “organokines”, “adipokines”, “hepatokines”, “myokines”, “osteokines”, “stellakines”, “fructose”, “gut microbiota”, “insulin resistance”, “crosstalk of organokines”, “obesity” and “type 2 diabetes (T2D)” from the last 10-years to date in 2023.
RESULTS
We found a total of 750 articles, out of which we selected 145 for this systematic review. No meta-analysis was performed.
PARTS OF INSULIN RESISTANCE, FRUCTOSE, GUT MICROBIOTA AND ORGANOKINES
Insulin Resistance, Inflammation besides Mitochondrial Impairment
IR possesses a key role in the pathophysiology of NAFLD and NASH. In general, insulin possesses a controlling function in hepatic metabolic events. Furthermore, it works on other peripheral cells implicated in glucose uptake, its storage (glucogenesis), and protein production, in addition to fatty acid generation. Conversely, it possesses a blocking action over catabolic events like lipolysis, glycogenolysis, proteolysis, and gluconeogenesis. Hence, resistance to its actions produces numerous inimical sequelae regarding the metabolism.33
In cases of increased intake of nutrients (comprised of carbohydrates and lipids, besides fast foods) in association with a lifestyle of decreased physical activity, enhanced energy ingestion gets stored in AT. AT absorbs and stores glucose and free atty acids (FFA), acting as a compensating mode to neutralize the probable toxic actions of these circulating nutrients, inclusive of adipocyte hypertrophy and hyperplasia. As a consequence of escalated nutrition, IR enhances the flow of FFA towards the liver by enhancing lipolysis and lipogenesis along with hampering esterification of FFA, forming an imbalance amongst generation and input vis-à-vis oxidation and exporting of hepatocellular fat.8,34
Therefore, this kind of imbalance is implicated in escalated intrahepatic FA accrual. This accrual possesses lipotoxicity in view of cells having the incapacity of secluding greater reactive lipid molecules, which results in their mitochondrial oxidation enzymatic system being overloaded with resultant mitochondrial injury, endoplasmic reticulum (ER) stress, and autophagy. Obesity correlated with NASH causes adipocyte hypertrophy and cell degeneration, along with demise.1,8,35
Hepatocyte demise is correlated with the liberation of damage-associated molecular patterns (DAMP) to the adjacent cells, which, via the inflammasome, get transformed into pro-inflammatory cytokines (interleukin-1 (IL-1) along with IL-18). On liberation, IL-1 causes the attraction of neutrophils. In addition to activating neutrophils, hepatic stellate cells (HSCs), along with IL-18 liberation, cause the attraction and activation of macrophages. The cause of these reactions is the stimulation of innate immunity by DAMPs via pattern recognition receptors (PRR), hence activating transcription factor nuclear factor κB (NFκB), which stimulates the pro-inflammatory effect in NASH. This
is followed by stepwise pro-inflammatory cytokines along with chemokine liberation like tumor necrosis factor-alpha (TNF-α) and IL-6, which ends in the generation of reactive oxygen species (ROS). This whole scenario, in addition to liver sinusoidal endothelial cells (LSECs), possesses a direct effect on fibrosis along with the provision of a milieu of chronic regeneration that facilitates chromosomal abnormalities resulting in the formation of HCC.8,36
The escalated quantities of ROS possess the capacity of initiating cell demise in hepatocytes by activation of a particular pathway, stimulating lipid peroxidation by compromising oxidation, leading to the decreased generation of reactive lipids. Such reactive molecules might further aggravate liver injury and facilitate the liberation of greater ROS outside hepatocytes, aiding in HSC activation along with extracellular matrix (ECM) getting deposited.35,37
Generation of malondialdehyde (MDA) takes place in the event of peroxidation, which is further implicated for perisinusoidal along with periportal fibrosis by activation of NFκB along with controlling the expression of pro-inflammatory cytokines like TNF-α along with IL-8. Such inflammatory biomarkers result in the activation of HSCs, which aids in fibrosis. Membrane phospholipid peroxidation changes their permeability and facilitates the ballooning of hepatocytes. This alteration might point to cytoskeletal injury with the incapacity of full programmed cell death (PCD). This alteration results in the accrual of hydrogen peroxide in hepatic peroxisomes, which, in combination with ferrous iron, generates free hydroxyl (OH) radicals that possess great reactivity with membrane phospholipids.35,38
Lipotoxicity is correlated with the transit of mitochondrial membrane pores that cause a break in cellular respiration, the production of OS, and the extravasation of cytochrome c from the cytosolic matrix, which activates the apoptosome in apoptosis. The escalated free radicals impact the mitochondrial deoxyribonucleic acid (mit DNA), resulting in decreased oxidative phosphorylation as well as depletion of adenosine triphosphate (ATP) stores. The hampering of electron flow in the phosphorylation chain produces greater ROS, which further injures mitochondrial DNA, resulting in a vicious cycle. Thus, apoptosis gets given up on depletion of ATP, replaced by necrosis or necroptosis taking over—a mode implicating mitochondrial injury, OS, along with activation of c-Jun-N-terminal kinase (JNK).35,39
Hence, variable pathological stimuli inclusive of hepatocyte demise and molecules liberated by AT along with intestinal pathogens might facilitate inflammation and fibrogenesis by activating Kupffer cells (KC) (alias resident liver macrophages). Obesity, along with KC, is a key part of the NASH pathophysiology. They facilitate the activation of M1-macrophages in contrast to the anti-inflammatory M2 macrophages. Activation of M1- macrophages generates different kinds of cytokines, which then enroll pro-inflammatory cells, thus exaggerating inflammation. These complicated sequences of processes finally end in the activation of HSCs, with subsequent escalated generation along with the deposition of ECM. Hence, depletion of adipocyte function in addition to inflammation of AT (existence of IL-1, IL-6, and TNF) aids in the generation of adipose resistance to insulin.12,23,38
Part of Fructose
Fructose possesses a noticeable role in NASH generation in view of its stimulation of hepatic fat accumulation and its correlation with MetS risk factors like hypertension, escalated serum triglycerides (TG), and IR. Noticeably, where fructose comes from sweetened fructose drinks apart from fruit ingestion, separate outcomes are obtained in view of the latter possessing lesser fructose quantities besides possessing antioxidants, which counteract the action of this monosaccharide.40
Entry of fructose takes place in hepatocytes through glucose transporter 2 (GLUT2), and at the cellular level, it gets transformed into fructose-1 phosphate (F1P) by fructokinase. Subsequently, aldolase B acts on F1P, generating phosphotriosis that can be transformed into glucose, lactate, and fatty acids. Following an acute fructose load, the lipogenic pathway that’s usually less active gets overactive on an escalation of phosphotriosis (Figure 2).38,41
Figure 2. Fructose Vias in Hepatocytes and their Relations with Fat Liver Accumulation
Adopted from Margiotti dos Santos et al11 increase,#: decrease, +: plus, ACC: Acetyl-CoA carboxylase, ChREBP: Carbohydrate-responsive element binding protein, ER: Endoplasmic reticulum, FASN: Fatty acid synthase, FIP: Fructose-1-phosphate, GLUT2: Glucose transporter 2, MT: Mitochondrial and SREBP1c: Sterol response element-binding protein 1c.
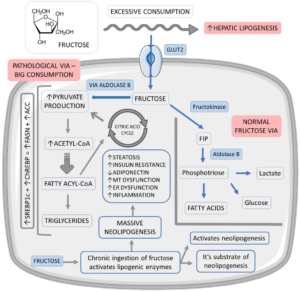
Additionally, decontrolling the hepatic entry as well as the metabolism of fructose ends in the escalated generation of acetyl CoA (glycolytic pathway) over the liver’s oxidative ability, facilitating neolipogenesis via the activation of factors like sterol regulatory element binding protein 1c (SREBP1c) and carbohydrate responsive element binding glucose protein (ChREBP). As a result of the activation of these factors, enhancement of fatty acid synthase (FASN) and acetyl CoA carboxylase (ACC) expression that controls lipid generation takes place. Saturation of the glycolytic pathway further facilitates the accrual of glycolytic intermediates that can get transformed into glycerol-3 phosphate that gets utilized in TG generation.41,42
Hence, fructose is the most robust lipogenic carbohydrate in the generation of hepatic steatosis in view of its being a substrate in addition to an activator of neolipogenesis. Chronic consumption of fructose escalates the events by stimulating the generation of lipogenic enzymes. Neolipogenesis stimulation, along with lipid accumulation in the liver, can result in hepatic IR. Furthermore, fructose has been acknowledged to hamper adiponectin generation and liberation, aiding in escalated IR and facilitating hepatic steatosis. These processes can facilitate OS in view of mitochondrial impairment and ER stress, factors that stimulate inflammation and propagate from simple steatosis to NASH.41,43
Fructose is immediately phosphorylated by fructokinase C without any negative feedback regulation. This event decreases the generation of ATP and results in its utilization in view of the fact that, whereas fructokinase phosphorylation is rapid, the cleavage response by aldolase B is comparatively slow.41 Hence, escalated fructose ingestion, which is more than advocated by the World Health Organization (WHO), which is 5% in contrast to the full advocated calories per day, can result in phosphate deficiency with the accrual of adenosine monophosphate (AMP) and sequentially escalated uric acid generation, with fructose reflecting the only common carbohydrate that generates uric acid in its metabolism.36,40 Activation of the enzyme AMP deaminase takes place subsequent to the reduction of intracellular phosphate, which transforms AMP into inosine monophosphate (IMP), leading to nucleotide renewal, which initiates the production of uric acid and aids in the generation of ROS.40,43
Noticeably, it is of considerable importance that, despite the fact that glucose and fructose are isomers and hence possess the same formula (C6H1206), the initiation of metabolism of these 2 molecules varies. The major variation is the implication of phosphofructokinase (a controlling enzyme) in glucose metabolism but not in fructose metabolism. This variation to start with results in reduced intracellular phosphate and ATP quantities, along with uric acid production, which is adequate for the blockade of protein generation, stimulation of ROS, and mitochondrial impairment leading to the MetS phenotype. Subsequent to this stage, the metabolism of both carbohydrates is similar.40,44 Uric acid accrual can interfere with fatty acid oxidation (FAO) and decrease hepatic ATP quantities with transient blockade of protein generation (OS), along with mitochondrial impairment induction. Additionally, uric acid possesses direct pro-inflammatory actions by activating NFκB and inducing inflammatory cytokine liberation. Lastly, fructose leads to the rupture of tight intercellular junctions, aiding in the escalated intestinal permeability that helps endotoxin entry via the portal vein, leading to NASH becoming indelible.40,43,44
Additionally, mitochondria possess enzymes possessing sensitivity to OS (acotinase 2 along with enoyl CoA hydratase). In combination, fructose and uric acid decrease the effect of acotinase 2, an enzyme correlated with Kreb’s Cycle that leads to the accrual of citrate. The escalated citrate is guided towards the cytoplasm, activating lipogenesis via stimulation of ATP citrate lyase. Moreover, it is noticeable that repeated sugar exposure led to glucose transport via GLUT5 as well as increased quantities of fructokinase in the liver, which escalated fructose absorption.40,45
Escalated along with chronic ingestion of fructose in view of a greater glycemic index results in an escalated need for ER stress in view of a greater lipid metabolism. Hence, endoplasmic reticulum membrane proteins might be fractalized or lead to lipid accumulation, aiding in ER stress, inflammation, OS, and apoptosis. ER stress further indirectly stimulates TG accrual in the liver, leading to hepatic and adipose IR, as well as activating transcription factors correlated with inflammation and cell demise. Fructose facilitates the generation of saturated fatty acids that possess the capacity to activate toll-like receptor 4 (TLR4) in the liver. Such activation stimulates OS secondary to the generation of inflammatory cytokines. Moreover, ER stress and inflammation can result in the generation of DAMPs, which signal and aid in alterations of metabolism. Escalated fructose can alter the profile of organokines, with escalated formation of fetuin-A, fibroblast growth factor 21 (FGF21), leukocyte cell-derived chemotaxin (LECT), and angiopoietin-like protein-8 (betatrophin/ ANGPTL8) implicated in alterations in energy homeostasis in IR and that can aid in organ injury.8,41,46
Role of Intestinal Microbiota
The liver obtains most of its intestinal blood flow via the portal system. Hence, it represents the initial line of defense regarding intestinally obtained toxins, along with exposure to numerous pathogen-associated molecular pattern receptors (PAMP): hence, the role of the gut microbiota (GM) in the pathophysiology of NASH is significant.47 Escalated consumption of foods rich in sugar and fat is correlated with the robust elimination of bacterial spp. variations and a remarkable decrease in the full density of the microbiota. Secondary to that, advantageous bacteria might be eliminated, besides the pathological microbiota getting overrepresented. Advantageous bacteria are inclusive of Lactobacillus, Bifidobacterium, Ruminococcus, Roseburia, Faecalibacterium and Bacteroides species (spp.) that possess anti-inflammatory actions besides being beneficial to the metabolic paradigm. Bacteria believed to damage the intestinal microbiota are Clostridium, Enterobacter, and Enterococcus spp. NASH patients have decreased quantities of Bacteroides in contrast to healthy subjects.48 Dysbiosis facilitates the transport of bacteria in addition to bacterial products (endotoxins) (like lipopolysaccharide (LPS) that might be correlated with the generation of fibrosis by activation of HSCs) to the portal circulation, hence aiding in the activation of HSCs in the propagation of NAFLD to NASH. Diet-induced obesity (DIO) changes the expression along with the organization of tight junctions amongst intestinal cells, which is directly correlated with escalated intestinal permeability. Moreover, NASH patients possess an increased capacity of ethanol-generating bacteria, in particular Escherichia, pointing out that ethanol-generating microbiota (Bacteroides, Bifidobacterium, and Clostridium) can work as a risk factor for NASH propagation. Hence, apart from pathologies that change the intestinal microbiota, its constituents are further impacted by diet.35,37,43,48
Here, fructose is a significant constituent. It stimulates the alterations of the intestinal microbiota, decreasing the expression of cell junction proteins as well as impacting cell adhesion. These events aid in the endotoxin’s entry via the portal vein, which stimulates the upregulation of lipogenic genes along with pro-inflammatory genes. The greater permeability aids in bacterial translocation from the intestine to the blood stream with the provision of signals to different organs, including the liver, a process that might stimulate the innate immune response along with escalating the pro-inflammatory reaction.35,43,49
Scientific proof pointed out that alterations in lifestyle, sticking to a diet and exercising, in addition to decreased body weight, led to an improvement in NAFLD. Escalated dietary fiber ingestion, probiotics, prebiotics, and calorie limitations are advocated. Dietary fiber is the most indigestible part of major diets, impacting the modulation of digestion in the form of a substrate for microbial fermentation. Moreover, the breakdown of fibers, which work in the form of prebiotics, yields numerous short-chain fatty acids (SCFA) that confer protection to the structure and function of the intestinal barrier. A diet possessing high fibers escalates the quantities of Bifidobacterium in addition to reducing the Firmicutes : Bacteroides ratio in humans, apart from decreasing the eating quantities in view of escalated satiety aiding in caloric limitations, a factor leading to improvement of NASH. Certain studies illustrated that greater ingestion of insoluble fibers (7.5 g daily) possesses the capacity to improve three separate liver fibrosis scores (hepatic fibrosis index, fatty liver index, and liver fat index NAFLD).35,48,50
Part of Diacylglycerol
Diacylglycerol (DAG) further possesses a key role in the pathophysiology of NASH, leading to hepatic IR and the propagation of liver steatosis. DAG is the second-last intermediate for TG generation and has been evaluated as the mediator of IR in hepatocytes by activation of protein kinase C epsilon kind (PKCε). PKCε phosphorylates the threonine residue on insulin receptors in hepatocytes, leading to dysfunctional insulin signaling in these cells and IR-obtained lipid depositing stimuli. Furthermore, in subjects with NAFLD or NASH, DAG expression in the liver is feasible in greater quantities, escalating TG generation remarkably. Escalated hepatic IR along with liver lipid generation is evaluated in the form of DAG-modulated liver fatty accrual along with steatohepatitis.51 Loomba et al52 assessed if antisense hampering of diacylglycerol-acetyl transferase (DGAT2) could efficaciously decrease liver fatting in subjects with diabetes and NAFLD and illustrated that hampering decreased liver fat quantities without resulting in hyperlipidemia.
ORGANOKINES
The endocrine workings of the liver, AT, and hepatic tissue are significant regarding NASH generation. The tissues possess the capacity to generate a biomarker peptide that has been acknowledged to be an organokine (hepatokines, adipokines, and myokines, respectively), which crosstalk via autocrine, endocrine, and paracrine pathways. The working of the organokines in combination is associated with health or the generation of variable diseases. With the escalation of AT, an escalation of the liberation of pro-inflammatory organokines aiding in metabolic conditions like IR, T2D and MetS takes place.14
The intricate association between skeletal muscle and bone tissue is much greater than simple anatomy; these tissues are further physiologically communicated by the endocrine system, which finishes the biochemical crosstalk. On chronically escalated energy ingestion, an escalated adiposity along with lipid accumulation in the bone marrow and adjacent myocytes leads to the liberation of FFA that possess lipotoxicity for muscle and bone cells located there. A vicious cycle gets triggered by these events, with chronic low-grade systemic inflammation (LGI) ensuing and leading to dysfunctional metabolism. At the time of resistance exercises, the load application to skeletal muscle gets switched to bone, which, apart from muscle protein generation, further signals a greater energy need for promoting bone generation. Certain systemic mediators, including leptin, can start muscle hypertrophy along with bone generation, validating that these tissues get endocrine signals along with bone and muscle having a bidirectional pathway for biochemical signals. These signals are brought about by myokines, osteokines, and adipokines that have autocrine, paracrine, and endocrine actions, hence controlling muscle as well as bone metabolism.53
In view of insulin being a generator of necessary hormones like insulin-like growth factor (IGF1), angiotensinogen, thrombopoietin, and hepcidin, the lipid imbalance secondary to hepatocyte injury and demise is further implicated in the decontrolling of the generation of organokines. There is a reduction in the expression of the insulin sensitizer and anti-inflammatory hormone adiponectin in NASH. Despite leptin being necessary for controlling the appetite in addition to escalating energy expenditure, its quantities are increased in obesity. Leptin facilitates fibrogenesis in stellate cells, which stimulates the generation of fibrogenic genes along with inflammation in T-cells.54
Various myokines like irisin, IL-6, IGF1, brain-derived neurotrophic factor (BDNF), and myostatin influence actions on bone metabolism, both anabolic and catabolic. Conversely, osteokines like osteocalcin and sclerostin stimulate muscle anabolism and catabolism, respectively. Adipokines like adiponectin, leptin, and resistin, along with TNF, can further impact bone and muscle anabolism. Moreover, lipolytic myokines, like Irisin, along with IL-6, are associated with exercise-stimulated thermogenesis as well as darkening adipocytes.53,55
Adipokines:
Apart from its part in energy storage, aiding in thermoregulation, and influencing mechanical protection, the AT further works in the form of an endocrine organ, liberating bioactive peptides la- beled as adipokines. These agents control lipid metabolism, apart from impacting insulin sensitivity, appetite, and fibrogenesis, along with fat accumulation in the liver. Their effect is directly associated with adipocyte hypertrophy, basically in obesity, and is correlated with mild inflammatory situations.55 Apart from being involved in the etiopathogenesis of certain metabolic diseases, they mediate bone turnover, bone mineral density, and skeletal muscle catabolism in aging diseases like sarcopenia. The accumulation of visceral fat is correlated with increased quantities of adipokines.53
Adiponectin: Adiponectin represents one of the adipocytokines that exists in large quantities, circulating in large amounts in the peripheral blood.24,31 Normally, adiponectin gets liberated by adipocytes and is present in a lot of isoforms (multimeric and monomeric forms possessing full length and globular sub-forms, along with certain oligomers) and facilitates insulin sensitivity in the liver, skeletal muscles, and AT that includes the ectopic components and confers protection to cardiac tissue. The greater the expression of adiponectin, the lesser the AT mass. These adipokines possess anti-inflammatory effects, escalate FAO, escalate glucose uptake in skeletal muscle, and hamper gluconeogenesis.53
Adiponectin gets generated just by AT, stimulates the liberation of anti-inflammatory cytokines (IL-10), leads to blockade of NFκB activation, and hampers the liberation of TNF-α as well as IL-6. Weight reduction stimulates its production.56-58 It is an anti-fibrotic, anti-inflammatory, and adipokine expressed by white in addition to brown adipocytes. Its decrease is directly associated with IR and glucose intolerance. A reduction in its quantities can be brought about by the escalation of pro-inflammatory cytokines. Apart from aiding in inflammation, these adipokines possess the properties of thermogenesis induction along with FAO in skeletal muscle as well as the liver.57,59 Furthermore, adiponectin possesses the capacity to induce HSC quiescence.60 Hence, adiponectin can be believed to be an adipokine that confers protection. There is an inverse association of its quantities with RI, with a lesser association in obese subjects and patients with RI, like those with T2D, NASH, and hypertension.56
Leptin: Leptin represents an adipocyte-formed hormone possessing a lot of functions, having its receptors broadly expressed in umpteen peripheral tissues along with the hypothalamus but none in AT.24,31
The basic function of leptin is to control body weight, food consumption behavior, and energy metabolism. In chronic low-grade inflammation along with obesity, leptin stimulates the generation of IL-6 and TNF-α along with decreased adiponectin. Escalated quantities of leptin lead to adiponectin resistance, resulting in restriction of muscle FAO and decreased lipolysis in AT, actions that get neutralized by exercise.56
Leptin is associated with food ingestion and energy expenditure, and its influence on NAFLD might be associated with IR or failure of the anti-steatotic effect. Its liberation gets escalated by IL-1 besides TNF-α.56,57 Leptin increases glucose along with FFA absorption from the skeletal muscle, in addition to decreasing the quantities of intrahepatic lipids in view of FAO. The increased quantities of leptin aid in the effects of pro-inflammatory cytokines. Moreover, leptin stimulates the proliferation of HSCs along with the generation of profibrogenic and pro-inflammatory factors. Greater leptin quantities have further been illustrated to be correlated with the propagation of NAFLD.57 Lastly, it possesses the capacity of influencing a pro-inflammatory effect in view of disrupted relaxation of nitric oxide (NO) associated vessels through escalated OS as well as by escalated expression of endothelin, hence enhancement of angiotensin II, that in turn escalates the generation of leptin that further stimulates the liberation of proinflammatory biomarkers (like TNF-α, IL-6, besides monocyte chemoattractant protein 1 (MCP1) receptor escalating the expression of adhesion molecules (like vascular adhesion molecules (VCAM-1) intercellular cell adhesion molecule (ICAM-1), along with E-selectin).56
Omentin: Omentin gets liberated from the AT of the omentum, which is a 34 KD protein implicated in controlling the adipocytes differentiation, maturation, energy metabolism, immune response, inflammation, and insulin sensitivity. Two homologous isoforms (Omentin 1 and Omentin 2) exist in the circulation, with Omentin 1 being the major isoform. Omentin 1 works with the utilization of AMPK, NFκB/MAP-Kinase (extracellular signal kinase1/2 (ERK1/2), JNK, as well as p38) signaling systems via which it exerts anti-proliferative, anti-inflammatory, anti-oxidative, and angiopoietic actions. Omentin escalates insulin sensitivity along with sequential glucose absorption.31 Its major circulating type is omentin-1 (alias intelectin, a peptide comprised of 313 amino acids). Patients with glucose intolerance and diabetes that are newly diagnosed possess decreased quantities, aiding in arterial calcification. Its quantities are inversely associated with hyperglycemia, chemerin quantities, and inflammation, besides insulin resistance (namely, factors that facilitate the propagation of NASH.61 It can function in the form of an antiatherosclerostic agent and confer protection against liver diseases.57
Resistin: Resistin represents a low molecular weight adipocytokine that aids in IR, inflammation, and OS.24,31 The basic biological actions of resistin get manifested via different molecular targets (like FFA transport protein 1, acetyl-coenzyme A (CoA)-carboxylase (ACC), and AMP-activated protein kinase (AMPK), CD36) and get influenced by the amelioration of glucose metabolism, hampering of FFA-oxidation, and uptake.31 Resistin gets expressed mainly by macrophages and gets liberated by them secondary to activation by pro-inflammatory cytokines. Resistin is associated with insulin resistance in obesity. It possesses a pro-inflammatory effect that stimulates the liberation of IL-12 and TNF-α.9,54,58 Furthermore, it stimulates angiogenesis along with skeletal muscle cell proliferation.9 It decreases the quantity of mitochondria and fat accumulation. Greater expression of resistin was observed in NASH in contrast to simple steatosis in control individuals.58,62 It gets generated by AT macrophages (ATM) along with monocytes, the induction of IR, angiogenesis, and skeletal muscle cell proliferation.63
Vaspin: Vaspin gets generated by visceral AT (VAT), believed to be a member of the serine protease inhibitor family, capable of hampering Kallikrein 7, a protease possessing the capacity to breakdown insulin. In view of these, it can cause an improvement in glucose intolerance and insulin sensitivity and decrease the generation of pro-inflammatory cytokines, conferring protection to vascular tissue from apoptosis. Greater serum quantities are associated with elderly obesity due to escalating insulin sensitivity and physical conditioning, along with lesser quantities of leptin.56,64
Visfatin: Visfatin represents an inflammatory adipokine enzyme (alias nicotinamide phosphoribosyl transferase and pre-B-cell colony enhancing factor) that possesses growth factor action that is implicated in the generation pathway of nicotinamide adenine dinucleotide (NAD+), hence sirtuins (SIRT) function. It gets liberated actively by both adipocytes and macrophages; it is also observed in the circulation and in the ECM, where it controls the OS, immune response, apoptosis, and inflammation via Sirt-1- based and mitogen-activated protein kinases (MAPKs)-associated pathways.31 Visfatin is generated basically by adipocytes along with macrophages in AT and, to a lesser degree, by hepatocytes along with neutrophils. It is implicated in the conversion of preadipocytes to adipocytes along with generation, apart from the storage of triacylglycerols in AT.65 It possesses immunomodulatory and vasodilatory characteristics in view of its ability to enhance the generation of NO. Conversely, it further displays proinflammatory effects, facilitating pancreatic β-cell impairment and leukocyte activation along with the generation of pro-inflammatory cytokines in AT, leading to the liberation of TNF-α, IL-6, vascular cell adhesion molecule 1 (VCAM-1), and IL-1β (factors that result in dysfunctional insulin signaling).57,58,61 Moreover, visfatin stimulates endothelial proliferation along with the growth of smooth muscle cell factors. Its quantities are increased in obesity, MetS, T2D, or cardiovascular disease (CVD).
Myokines
Myokines Skeletal muscle is implicated in the liberation of myokines which can impact metabolic actions. This liberation is directly associated with continuous physical exercise. These organokines display how the muscle possesses the key liberating function, liberating peptides, and cytokines, in addition to growth factors. The kind of myokines liberated is associated with physical activity or lack along with the kind of muscle fiber as well as an exercise conducted.57
Irisin: Irisin, a proteolytic hormone derivative (myokine) of the fibronectin III domain-containing 5 (FNDC5) gene, has been demonstrated in a mouse model to be induced by exercise (mediated by peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) and to then directly stimulate phenotypic changes in AT, which mediates changes in systemic metabolism. This irisin preferentially acts on the subcutaneous “beige” fat and causes it to brown by increasing the expression of uncoupling protein 1 (UCP-1) and other thermogenic genes.15 Irisin is associated with the control of energy homeostasis and metabolism, along with crosstalk amongst skeletal muscle as well as other tissues. The differentiation of white adipose tissue (WAT) in addition to brown adipose tissue (BAT) induction is one of its functions. This differentiation to BAT results in repression of adipogenesis, cholesterol formation, and the maximization of lipid oxidation, thus maximizing lipid homeostasis. In general, it gets liberated subsequent to physical exercise, and apparently it escalates insulin sensitivity in view of stimulation of the mobilization of glucose transporters in insulin-based tissue, leading to enhancement of MetS as well as CVD. Apart from anabolic actions on bone, it escalates the generation of osteoblasts.53,60,65
Polyzos et al64 illustrated those greater quantities of this particular myokine existed in lean patients in contrast to obese patients as well as patients with NAFLD/NASH. Nevertheless, what was paradoxical regarding this study was that on portal inflammation evolution, an enhancement of irisin quantities might take place, which might be probably for restriction of inflammation as a compensating mode.64
Mionectin: Mionectin gets liberated at the time of resistance exercises and escalates lipid uptake by AT along with the liver, decreasing quantities of plasma FFA. Reduction in physical activity decreases its quantities, escalating the quantities of FFA in the blood and resulting in FA accrual in other tissues, producing IR.66Moreover, it influences FFA absorption by cells, apart from coordinating metabolic events in different tissues.66
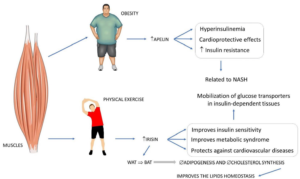
Myostatin: Myostatin promotes muscle depletion by hampering tissue growth, stimulating tissue breakdown, and facilitating muscle atrophy. Decreased muscle mass escalates metabolic conditions. The skeletal muscle works as a major source of glucose-based uptake; loss of this mode results in compensation in other organs like the liver. Once exercise is continued regularly, its quantities decrease; hence, physical activity possesses a significant role in the form of a therapeutic target. Its quantities are greater in obese patients and are intricately associated with IR, facilitating the downregulation of GLUT4 expression. It escalates the pro-inflammatory milieu and hampers the liberation of antioxidants and anti-inflammatory agents. Its decrease is directly associated with decreased resistive index (RI), lesser fat accrual, producing muscle hypertrophy with control of oxidative metabolism of skeletal muscles, lipolysis in WAT along with darkening of adipocytes, reorganization of FFA, and utilization in lipogenesis for getting used for fuel (factors leading to the avoidance of the propagation of NASH) (Figure 3 for exercise, the liberation of myokines, and NASH).57,67
Figure 3. Some Myokines and their Relations to Key Factors involved in NASH
Adopted from Margiotti dos Santos et al11 increase, #: decrease,Æ: restriction, WAT: white adipose tissue, BAT: brown adipose tissue
.
Hepatokines
Hepatokines possess a necessary role in modulating metabolic events and pathological situations, basically in the form of liverobtained pro-inflammatory factors.65 Fetuin-A, fibroblast growth factor 21 (FGF21), selenoprotein P, sex hormone binding globulin (SHBG), angiopoietin-like protein-8 (betatrophin/ANGPTL8), and leukocyte cell-derived chemotaxin-2 (LECT2) are the ones that have been evaluated the most to date. The liver’s capacity to influence glucose as well as metabolism by liberating these organokines into the blood, along with NAFLD, is apparently correlated with their changed formation. Moreover, hepatokines can be believed to be biomarkers of ectopic fat accumulation in the liver and markers of disease propagation. Certain hepatokines might be the target for avoidance along with therapy of diseases correlated with IR, including NAFLD.54,68
Angiopoietin-Like Protein-8: Angiopoietin-Like Protein-8 (AN- GPTL8/betatrophin) represents a circulating hepatokine also called TD26 and lipasin. It is significantly expressed in the liver and VAT. Overexpression of ANGPTL8 in BAT escalates lipoprotein lipase (LPL) action along with TG uptake. Serum ANGPTL8 has significantly escalated in patients with pre-diabetes and T2D.31 ANGPTL8 liberation by the liver takes place at the time of exercise. In view of it hampering pancreatic lipases, it hampers fat absorption. In the case of WAT, it stimulates lipolysis in addition to decreasing its growth. Its escalation is directly associated with IR as well as MetS.57 Apart from that, it plays a part in the reorganization of FFA in skeletal muscles instead of storage in AT. There is a reduction in ANGPTL8 in obese subjects. Hence, the hampering of LPL gets influenced, leading to lipid accrual along with the probable evolution of NASH.67,69
Leukocyte cell-derived chemotaxin2: Leukocyte cell-derived chemotaxin2 (LECT2) represents a neutrophil chemotactic organokine directly associated with metabolic stress in view of its leading to dysfunctional insulin signal transduction along with escalated inflammatory cytokines and adhesion molecules; escalated insulin sensitivity results from its depletion.57
Additionally, it can pick up hepatic lipid steatosis as well as positively control itself via the activation of lipopolysaccharide (LPS) signaling in macrophages prior to weight accrual. Its expression is directly associated with escalated nutrition as well as, in turn, BMI along with liver inflammation in parallel with NASH generation.70
Sex hormone binding globulin: Sex hormone binding globulin (SHBG) represents a protein that is basically liberated by the liver with the transportation of sex steroids to the target tissues. A reduction of SHBG takes place in T2D, obese subjects, and patients with hepatic liver steatosis, in contrast to subjects with no liver pathological conditions. The decreased expression of SHBG in NAFLD might take place in view of inflammation, which, subsequent to TNF-α escalation in reaction to JNK and NFκB activation, decreased its generation. Regarding metabolism, overexpression of SHBG confers protection against NAFLD by hampering hepatic lipogenesis via the downregulation of crucial lipogenic enzymes. Lifestyle manipulations, via obesity therapy as well as weight loss, result in increased circulating SHBG. Moreover, adiponectin escalated SHBG generation by activating the AMPactivated protein kinase (AMPK) and decreasing the action of enzymes implicated in liver lipogenesis. That is, with the reduction of quantities of hepatic SHBG in view of decreased hepatic generation due to escalated pro-inflammatory cytokines, hepatic lipogenesis gets facilitated. Hence, fat accumulation in the liver would decrease hepatic SHBG generation, which in turn would facilitate liver lipogenesis; hence, a vicious cycle gets triggered, which aggravates the generation of NAFLD.57,71
On liver injury, SHBG generation gets interfered with like with other sex hormones. Hence, its quantities are decreased in steatosis, obesity, and T2D. In the case of PCOS, greater quantities of androgens are implicated in the reduction of their generation, which facilitates the initiation or propagation of liver diseases, including NASH and metabolic diseases.72
Osteokines
The liberation of osteokines takes place by direct stimulation of osteocytes and osteoblasts by liberation of muscle matrix, producing a secondary reaction in the bone and other tissues with autocrine, endocrine, and paracrine effects. These osteokines possess systemic actions, with certain of them being osteocalcin (OCN) and BMP-7. OCN, sclerostin, and BMP are actively liberated by osteoblasts and can be liberated into the matrix at the time of bone resorption. Moreover, they possess considerably important actions on metabolism.56,67
Bone morphogenetic protein: BMP represents a member of the TGF-β family, controlling variable events like organogenesis and paradigms of embryonic generation in addition to white and brown adipocyte generation. Expression of its sub-kinds in AT, along with its quantities in the blood, are associated with obesity and fat redistribution. BMP-7 is implicated in appetite control, and a reduction in food ingestion is proportional to the degree of its greater quantities. Moreover, the BMP-4 sub-kinds are liberated by adipocytes, leading to the stimulation of adipogenesis and guiding preadipocytes to a brown adipocyte phenotype. At the time of obesity, preadipocytes are resistant to these BMP actions, which might aid in obesity-associated diseases like NASH.56
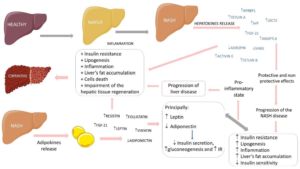
Moreover, BMP-7 stimulates the liberation of insulin by the pancreas. Hence, in view of its part in controlling food ingestion, energy expenditure, and AT formation, this class is necessary for the relief of obesity in addition to its associated co-morbidities (Figure 4 regarding the correlation of hepatokines along with adipokines in the propagation of fatty liver diseases).53,74
Figure 4. Relations between Hepatokines and Adipokines in the Progression of Fatty Liver Diseases
Adopted from Margiotti dos Santos et al11 increase #: decrease, +: plus, HFREP1: Hepatocyte-derived fibrinogen-related protein 1, SeP: Selenoprotein-P, LECT2: Leukocyte cell-derived chemotaxin 2, FGF-21: Fibroblast growth factor 21, ANGPTL4: Angiopoietin-like 4, SHBG: Sexual hormone binding globulin, IR: Insulin resistance, NASH: Non-alcoholic fatty liver disease and NAFLD: Non-alcoholic steatohepatitis.
Osteocalcin: OCN gets generated and liberated basically by osteoblasts, followed by activation by osteoclasts in bone resorption.75 Its uncarboxylated and subcarboxylated (unOCN) kinds escalate insulin sensitivity and liberation by direct stimuli in the pancreas. Subsequent to exercise, unOCN quantities enhance, hence stimulating glucose uptake along with escalating insulin sensitivity, which is also associated with muscle hypertrophy and power. Uncarboxylated OCN stimulates glucose uptake and escalates insulin sensitivity and IL-6 sensitivity in skeletal muscles in rats; however, its effects are doubtful in humans.53 OCN quantities are inversely associated with body mass index (BMI), IR, and inflammatory biomarkers, along with body fat mass. It facilitates the working and survival of pancreatic β-cells, enhancing insulin liberation, which by itself stimulates the liberation of OCN, hence generating a direct endocrine association between the bone and the pancreas. Moreover, it possesses advantageous actions in view of stimulating the generation of adiponectin along with IL-10. Conversely, it decreases TNF-α quantities in adipocytes and positively controls thermogenesis in AT and mitochondrial generation in skeletal muscles. Apart from that, it enhances glucose as well as FFA uptake by muscles.67,75
Furthermore, OCN possesses an association with the nervous system. If it is lacking, the hippocampus is smaller and under-generated, which results in memory dysfunction, whereas its existence would be associated with a reduction in age-associated decreased cognition along with aid in reaction to acute stress, hampering the parasympathetic nervous system.76-78 Hence, OCN stimulates the ingestion of glucose along with FFA by facilitating the expression of fatty acid transporters and stimulating β-oxidation along with GLUT4 translocation to the plasma membrane. Hence, an anti-inflammatory milieu aids in decreasing visceral fat and decreasing NASH (Figure 5 illustrates the major organokines implicated in the formation of NASH).7,75,76
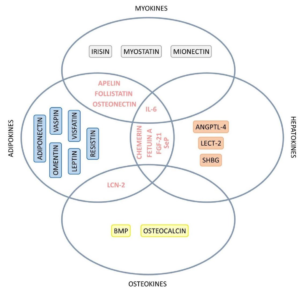
Figure 5. Main Adipokines, Hepatokines, Myokines and Osteokines involved in the Pathophysiology of NASH
Adopted from Margiotti dos Santos et al11 NAFLD: Non-alcoholic fatty liver disease, NASH: Non-alcoholic steatohepatitis, “: increase, #: decrease, F: impairment, +: plus, ANGPTL-4: angiopoietin-like 4, IR: insulin resistance, LECT-2: leukocyte cell-derived chemotaxin 2, SHBG: sexual hormone binding globulin, CVD: cardiovascular diseases, BMP: bone morphogenetic protein, β: beta, TNF-α: tumor necrosis factor alfa, IL-10: interleukin 10, TG: triglycerides, HSCs: hepatic stellate cells.
Crosstalk Amongst Organokines
Apelin: Apelin, an adipocyte-derived hormone, was reported to promote brown adipocyte differentiation by apelin-AP1 signaling, both by increasing brown adipogenic and thermogenic transcriptional factors via the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) and AMPK signaling pathways. They further reported that apelin relieves the TNF-α inhibition on brown adipogenesis and increases brown adipocyte activity, as shown by increased PGC-1α and UCP-1 expression, mitochondrial biogenesis, and oxygen consumption.15 Apelin is formed by fat tissue and muscle, and in obesity, its reduction takes place. Moreover, its expression takes place in the central nervous system (CNS), hypothalamus, heart, and stomach. It possesses a significant anti-inflammatory component, aiding in the regulation of heart muscle and, in turn, blood pressure (BP), besides cell cycle control and demise. This organokine further stimulates muscle regeneration, avoiding muscle depletion.58,79
Furthermore, it controls glucose metabolism, lipolysis, and cardiovascular homeostasis, decreasing lipolysis along with vasodilation induction, which facilitates the decrease in BP. Conversely, greater circulating quantities point to apelin resistance alone and are seen in obesity besides IR.56
Chemerin: Chemerin is believed to be an adipokine along with a hepatokine. Its expression takes place basically in WAT and gets liberated for controlling glucose uptake, lipolysis, and adipocyte differentiation. Its activation takes place by inflammatory and coagulation proteases.58,80 Apart from that, it is believed to be a chemoattractant for dendritic cells and macrophages. Additionally, it controls bone metabolism via testosterone (T) generation along with a balance between osteoblasts and osteoclasts.80
Chemerin escalates glucose tolerance and hampers insulin signaling, along with possessing a part in bone breakdown besides its generation and stimulating inflammation.57,80 In view of the association between obesity and inflammation, it is escalated in NASH. Its quantities are directly correlated with components of MetS like escalated BMI, hypertension, escalated triglycerides (TG), and C-reactive protein (CRP) quantities. Chemerin quantities are elevated in obese as well as T2D subjects, with it possessing an inverse association with high-density lipoproteincholesterol (HDL-C) along with omentin quantities.61,81,82
Fetuin A: Fetuin-A is one of the liberated glycoproteins believed to be the first hepatokine demonstrated to be correlated with metabolic diseases. Fetuin-A is positively correlated with hepatic steatosis and IR.1 Its amounts are increased in patients with NAFLD, NASH, and T2DM. In the form of a significant source of NAFLD generation, FFA escalates pro-inflammatory fetuinA expression. FFA-stimulated Fetuin A works as an endogenous ligand of TLR4 and accelerates lipid-modulated IR.31 Fetuin-A reflects an adipokine along with a hepatokine, and its escalated quantities are correlated with obesity, IR, T2D, and NAFLD, along with hampering ectopic calcium getting deposited.8,24 Fetuin-A facilitates the liberation of pro-inflammatory biomarkers in monocytes and adipocytes, along with downregulating the expression of adiponectin, an insulin-sensitizing hormone.54,56,65,68
It naturally hampers insulin receptors in the liver and skeletal muscles, facilitating pro-adipogenic actions in view of its greater quantities that disturb the insulin signaling steps along with GLUT4 translocation.56.57 Hepatocytes liberate Fetuin-A in the blood, which binds to insulin receptors in tissues, hampering their signaling, in view of which IR results.68 Moreover, Fetuin-A stimulates the liberation of inflammatory cytokines in monocytes, besides AT. Reduction in Fetuin-A results in advantageous actions in view of decreased accessible FFA, reducing visceral AT mass.57
Fibroblast growth factor: FGF19 as well as FGF21 belong to the FGF19 family, which needs the Klotho proteins as cofactors. They stimulate FGFR4 along with Klotho, which has an abundance of expression in hepatocytes. FGF19 and FGF21 have the role of controlling glucose, lipid, and bile acid metabolism.31 FGF21 liberation takes place in AT, and the liver is a controller of glucose in addition to lipid metabolism. Its escalated quantities in NAFLD are associated with quantities of hepatic triglycerides; hence, FGF21 is believed to be an upcoming biomarker of NAFLD. It might be a probable therapeutic agent for tackling IR in view of its resulting improvement of insulin sensitivity, body weight, lipid profile, and hepatic IR, along with its potential role in inflammatory manipulation.1 Furthermore, it stimulates the darkening of WAT.58 In view of it stimulating the expression of thermogenic genes as well as adiponectin and FAO in the liver, along with insulin sensitivity. FGF21 escalates FAO in the liver, decreasing glucose generation, thus sequentially postponing steatohepatitis generation. It further escalates glucose retention in adipocytes in view of escalated GLUT1 expression, hence an improvement in insulin sensitivity. On FGF21 liberation into muscle, in reaction to insulin and escalated glucose uptake, it might stimulate bone resorption.53,54,57,65,68,80
Follistatin: Follistatin reflects an adipokine as well as a myokine; it is liberated at the time of exercise, leading to the induction of thermogenic genes along with neutralizing myostatin.66
Follistatin is basically liberated by the liver, and its quantities are escalated with an escalated glucagon: insulin ratio, like in fasting besides exercise. Patients presenting with IR secondary to obesity possess greater quantities of Follistatin. In view of its capacity to neutralize myostatin, it can escalate skeletal muscle hypertrophy apart from inducing thermogenic genes in murine adipocytes. Its continuously escalated quantities are associated with IR, escalated hepatic glucose generation, and glucose intolerance. Conversely, temporary enhancement subsequent to physical activity is advantageous in view of it facilitating glucose as well as FFA uptake by skeletal muscle cells along with lipolysis in WAT.67
IL-6: IL-6, a pro-inflammatory and prooncogenic cytokine, is believed to be an adipokine, a hepatokine, and a myokine. It is an anticipatory biomarker for IR as well as CVD. It represses adiponectin quantities along with those of TNF-α and stimulates leptin generation.9,66 The generation of IL-6 takes place in macrophages, which are stimulated by NFκB activation. Hampering of insulin receptor substrate-1 (IRS-1), in addition to GLUT-4, in adipocytes.66 Whereas IL-6 obtained from AT stimulates inflammation and glucose intolerance along with bone resorption, IL-6 obtained from myocytes possesses an anti-inflammatory effect in view of the fact that it liberates IL-10, generating an antiinflammatory milieu, escalating glucose uptake, lipolysis in WAT, darkening of adipocytes, and metabolism improvement.53,57,67,80
Lipocalin 2: Lipocalin 2 (LCN-2) (neutrophil gelatinase-associated lipocalin) gets liberated by different cell types belonging to the lipocalin protein superfamily. A broad expression occurs in AT, resulting in inflammation in addition to fibrosis. There is proof with regards to overexpression of LCN-2 in WAT getting regulated by upregulation of IL-1β.31 LCN-2, initially believed to be an adipokine, was later observed to be an osteokine too. Its expression takes place in osteoblasts along with adipocytes and aids in a feeling of satiety and energy expenditure. Obese subjects might possess resistance to LCN-2, which escalates with aging and results in a reduction in energy expenditure.53,57 LCN-2 quantities were observed to escalate in animal models of diet-induced NASH liver injury and inflammation. This alteration was correlated with enhanced intrahepatic neutrophils along with the generation of pro-inflammatory cytokines that aided in the sustenance of inflammatory events.82 Hence, LCN-2 works as an inflammatory biomarker that is correlated with infections, ischemia, and renal inflammation, along with conditions like T2D and MetS.83
Osteonectin: Osteonectin has been seen to be an adipokine and a myokine (alias secreted protein acidic and rich in cysteine (SPARC)). The expression of pro-inflammatory cytokines implicated in IR and adipogenesis stimulates insulin liberation along with erythropoiesis, which possesses a significant role in the avoidance of mitotic clonal expansion of preadipocytes, hampering the start of adipogenesis. In view of the hampering of adipocyte generation, it limits lipid storage, escalating circulating quantities and resulting in hyperlipidemia, followed by fat accumulation in the liver and skeletal muscle.57
Mazzolini et al84 found lesser quantities of SPARC to confer protection against NASH apart from decreased liver injury in robustly obese subjects. Consequently, its lack has been associated with changes in hepatic lipid metabolism and the chances of the generation of HCC, which is intricately associated with NAFLD.85 The decreased quantities of FGF21 in adult patients with robust liver steatosis are in view of damage or demise secondary to lipotoxicity as well as hepatic inflammation. This decrease might further be correlated with disease propagation in NASH. The escalated FGF21 generation might be a probable factor that confers protection against carbohydrate as well as lipid metabolism conditions in view of SPARC directly controlling lipid metabolism and hepatic accrual in an insulin-independent way. Furthermore, it disrupts different paradigms of the “multi-hit” model of NAFLD like mitochondrial impairment, OS, chronic low-grade inflammation, and ER stress.86
Selenoprotein P: Selenoprotein P (SeP) gets generated by the liver along with AT, aiding in the generation of IR as well as T2D.7,75
It functions in the form of intra-cellular antioxidants in phagocytes, reducing inflammatory reactions via the differentiation of macrophages from M1 to M2, decreasing oxidative injury, and hence avoiding injury to hepatic endothelial cells.
Nevertheless, its role has been contradictory regarding their quantities at the time of the inflammatory reaction, inclusive of metabolic diseases as well as the existence of NAFLD and NASH.56,57,64,67
TNF-α: TNF-α is a cytokine that aids in steatosis, OS, and is an inflammatory mediator cytokine liberated by multiple inflammatory tissues apart from being an inducer of hepatocyte demise. Escalated quantities of TNF-α are correlated with obesity and IR via upregulation of suppressor of cytokines signaling 3 (SOCS3).76 Hepatocytes contain TNF superfamily death receptors that get activated at the time of propagation from NAFLD to NASH. On binding with TNF receptor-1 (TNFR1), induction of activation of caspase-3 and caspase-8 takes place, resulting in apoptosis of liver cells, a significant part in the breakdown of injured cellular constituents. At times of stress like inflammatory liver disease, apoptosis takes place for cell conservation; however, escalated apoptotic events might result in irreversible cell atrophy and its collapse, further aiding in the propagation of NASH.77 TNF-α escalates lipolysis, hence escalating FFA quantities and facilitating the generation of IR. Moreover, by stimulating and activating vascular adhesion molecules, it promotes atherogenesis. The other mode implicated in TNF-α induction of IR in peripheral tissues is the activation of NF-B and stimulating cytokines along with adhesion molecules. Moreover, it works as a chemoattractant for monocytes and neutrophils. It escalates the cytotoxicity of monocytes and macrophages, acting simultaneously as a cytotoxicity mediator. The maximum of its functions get mediated by its capacity for the formation of other cytokines functionally correlated with TNF-α, ECM proteins, monocyte modulation, and fibroblast chemotaxis, apart from the expression of vascular adhesion molecules. Hence, in view of its multiple pro-inflammatory actions, it aids in NASH evolution.61
Transforming growth factor-beta: Transforming growth factorbeta (TGF-β) represents a pleiotropic cytokine implicated in proliferation, survival, differentiation, angiogenesis, and wound repair reactions. Certain studies pointed out that hepatocytes with escalated lipids escalate the generation of ROS modulated by TGF-β leading to the hepatocytes demise. Hence TGF-β signaling in hepatocytes aids in the hepatocytes’ lipid accumulation, along with the generation of ROS, which facilitates the generation of NASH. In the liver, TGF-β signaling takes part in fibrogenic reactions via the activation of HSCs. Hence, TGF-β signaling in HSCs plays a significant role in the propagation of fibrosis in the case of advanced NAFLD (Figure 6 sums up the roles of different hepatokines implicated in the pathogenesis of NASH).78
Figure 6. Venn Diagram Showing the Organokines with Simultaneous Classification involved in the Pathophysiology of NASH
Adopted from Margiotti dos Santos et al11 IL-6: Interleukin 6, FGF-21: Fibroblast growth factor 21, SeP: Selenoprotein-P, ANGPTL-4: Angiopoietin-like 4, LECT-2: Leukocyte cell-derived chemotaxin 2, SHBG: Sexual hormone binding globulin, LCN-2: Lipocalin 2, BMP: Bone morphogenetic protein.
Stellakines
The activation of HSCs has a significant association with the propagation of chronic liver diseases, including liver cirrhosis and liver cancer. Activated HSCs liberate multiple pro-inflammatory factors and generate ECM constituents like α-SMA and type I and type III collagen fibers, which guide liver inflammation and manipulate hepatocyte demise, hence facilitating the generation of liver disease.87 Xiong et al88 observed that activated HSCs in the NASH mouse model, apart from forming ECM constituents, liberated abundant signal protein molecules labeled as “stellakines”, like i) amyloid precursor protein (APP), ii) CC chemokines (like CCL2, CCL11), iii) macrophage-colony stimulating factor (MCSF, alias CSF1), iv) connective tissue growth factor (CTGF, also called CCN2), and CXC chemokines (such as CXCL1, CXCL10) (Figure-7).
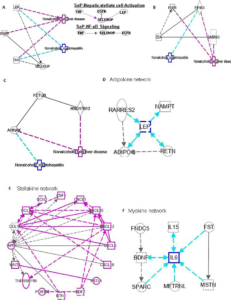
Figure 7. Ingenuity Pathway Analysis (IPA) of SELENOP, EDA and FETUB. (A) IPA investigated the Molecular Pathways of SELENOP
involved in NAFLD and NASH
Adopted from Yang et al142 All identified pathways of SELENOP were screened individually. Representative canonical pathways of SELENOP are associated with other related genes of NAFLD and NASH. Functional relationships of these genes and SELENOP are depicted using straight lines with arrows. SELENOP is functionally related to 1. Hepatic stellate cell signaling and 2. NF-κB signaling (Right). (B, C) IPA investigated the molecular pathways of EDA and FETUB involved in NAFLD and NASH. All identified pathways of EDA and FETUB were screened individually. (D-F) IPA investigated the molecular pathways of Adipokine, Stellakine and Myokine network.
CSF1 along with periostin: CSF1 reflects a myeloid cytokine liberated at the time of infection and inflammation, which has further been illustrated to control the differentiation, proliferation, survival, and activation of monocytes and macrophages.89 Mohallem et al90 illustrated that colony stimulating factor receptor 1 (CSF-1R/CSF1) is considerably upregulated in cells treated with TNFα-, pointing out that upregulation of CSF1 might activate pro-inflammatory reactions and leukocyte invasion of AT via non-classical activation of NF-κB signaling modulated by the TNF receptor. The non-classical NF-κB, the p52-RelB dimer, is believed to react to selective receptor signals that mediate adaptive immune functions. NF-κB-inducing kinase (NIK) is a central constituent in the non-classical NF-κB signaling pathway.91 Studies have further illustrated that the CSF1R facilitates the induction of non-classical NF-κB signaling at the time of macrophage differentiation,92 which might possess the probability of hepatic healing in the case of fibrotic livers since macrophage accrual and phagocytic activity aid significantly in this event.93 Periostin (POSTN) (another stellakine) is basically obtained from activated HSCs, and its main function is controlling cell adhesion, proliferation, migration, and apoptosis.94 POSTN expression can be induced by NF-κB and other pro-inflammatory transcription factors in vitro. 80,95 Nevertheless, POSTN cooperates with TNF-α or IL-1α to activate NF-κB in fibroblasts and then induces the expression of CCL2/MCP1, CCL4/MCP1, CCL7/MCP3, CXCL1/KC, CXCL2/MIP-1α, and IL-1β.96 These results show that crosstalk between POSTN and NF-κB activation might be a crucial factor in controlling the inflammatory pathogenic association of NAFLD.
NTN1, GAS6 along with WNT4y: Netrin-1 (NTN1) is a member of the family of laminin-associated liberated proteins. NTN1 controls the activation of NF-κB via the uncoordinated-phenotype-5A (alias UNC5A, an NTN1 receptor), and apart from the activation of UNC5A significantly, it activates the phosphorylation of NF-κB/p65 at Ser536 however, UNC5A upregulates the expression of c-MYC (the NF-κB downstream target) by activating NF-κB to facilitate cell proliferation.97 The RAC1 gene is implicated in cellular growth and cell-cycle control. KCNQ1 opposite strand/antisense transcript 1 (KCNQ1OT1) is a long noncoding ribonucleic acid (RNA) gene. Interference or hampering of NTN1-NF-κB further hampers cellular myelocytomatosis oncogene (c-MYC), resulting in downregulation of the expression of RAC1 and hampering of the hedgehog pathway through downregulation of KCNQ1OT1, finally hampering HSC proliferation and epithelial-mesenchymal transition (EMT) in liver fibrosis.98
Growth arrest-specific 6 (GAS6) is believed to be implicated in the stimulation of cell proliferation. GAS6 hampers NF-κB phosphorylation, chemotaxis, and adhesion affinity amongst monocytes and endothelial cells.99 GAS6 avoids activation of TNF receptor-associated factor 6 (TRAF6) and NF-κB by upregulating the suppressor of cytokine signaling 3 (SOCS3) in cellular models.100 AS6-AS2 is an identified cancer-related lncRNA. TLR ligands reduce GAS6 by downregulating GAS6-AS2 expression via NF-κB activation, pointing to a bidirectional feedback system between GAS6 and inflammation.84 WNT4 hammers NF-κB by non-classical WNT signaling.101 WNT4 further activates β-catenin/NF-κB by the working together of frizzled 4 and low-density lipoprotein receptor-related protein 6 (LRP6) signaling.102 Recombinant WNT4 proteins robustly hamper the apical step of TGF-β-activated kinase 1 phosphorylation, along with the following steps of receptor activator of nuclear factor kappa beta (NFkB ligand) (RANKL) (RANKL induces each critical step in NF-κB activation)-induced p65 phosphorylation and IκBα phosphorylation and breakdown.101,103 Moreover, WNT4 further hampers NF-κB-based transcription.101 These facts further illustrate that various genes crosstalk with NF-κB to control the generation and propagation of NAFLD.
CXCL1, CXCR4/7 along with CXCL16: CXC chemokines are a chemokine subfamily with a CXC motif at the N-terminus, also called α-chemokines. Seventeen (17) CXC chemokines have been displayed (like CXCL1, CXCL10, CXCL12, CXCL14, and CXCL16). CXC chemokines impact tumorigenesis on cell enrollment, collection, migration, invasion, angiogenesis, angiostasis, and lymphangiogenesis.104 CXCL1 has an extra ELR motif, and VEGF activity induces escalated expression of CXCL1 in endothelial cells, which induces angiogenesis via CXCR2 (the receptor of CXCL1).105,106 Activation of hypoxia-inducible factor-1 (HIF1) and NF-κB escalates CXCR2 expression in cancer cells at the time of chronic hypoxia.107 In human umbilical vein endothelial cells (HUVECs), NF-κB/p65 escalates the expression of long intergenic non-protein-coding RNA 1693 (LINC01693, which works as a miRNA-302d sponge) to escalate CXCL12 expression.108 miRNA-302d reduces the expression of CXCL12, which illustrates that hypoxia enhances the expression of CXCL12 via NF-κB.104 Mallory-Denk bodies formed in human alcoholic hepatitis and NASH can mediate TLR3/4 signaling through the NF-κB-CXCR4/7 pathway.109 LPS and NF-κB activation increase the expression of CXCL16 in HUVEC.110 Silencing of CXCL16 expression decreases tumor cell migration and proliferation via decreasing NF-κB activation.111
CCL11, CCL2, CXCL10 along with CTGF: CC chemokines are a subfamily of 27 chemokines that are implicated in intercellular connection and can further control the microenvironment in certain tumors.112Activation of ERK MAPK by CCL11 mediates apoptosis resistance in cancer cells.113 CCL11-CCR3 crosstalk activates the phosphatidylinositol 3-OH kinase/serine-threonine kinase (PI3K/Akt) signaling pathway in endothelial cells to induce angiogenesis.114 Moreover, activation of the CCR3 receptor by CCL11 escalates the expression of vascular endothelial growth factor (VEGF) in HCC, thus indirectly facilitating angiogenesis.115 It has been reported that macrophages with NF-κB-mediated mixed canonical and signal transducer and activator of transcription (STAT)-6-mediated alternating activation phenotypes can generate CCL11.116 This incites the thought that NF-κB regulation and activation of CCL11 can facilitate cancer angiogenesis and affect the prognosis of HCC. p50 can hamper liver inflammation and immune response by controlling CCL2 and CXCL10 liberation by HSCs, attenuating liver injury.117 p65 inhibits the activation of HSCs and reduces the liberation of stellakines in the inflammatory response.118 Tumor cell-derived connective tissue growth factors (CTGF) transmit growth-promoting signals to HCC cells by activating nearby HSCs, so CTGF has also been identified as a cornerstone in the HCC microenvironment, but anti-CTGF antibodies are prone to hampering this crosstalk.119CTGF facilitates the nuclear accumulation of p50 and p65 to protect the survival of the primary HSCs. Chaqour et al118 illustrated that NF-κB binds to the binding region in the promoter area of CTGF. Intranuclear translocation of NF-κB via the Smad independent pathway can control CTGF expression, while TGF- -induced CTGF expression is hampered by Bay 11-7082 (NF-κB inhibitor).119
NAFLD AS WELL AS NASH-OBTAINED HCC
Pathogenesis of NAFLD and NASH-associated HCC
HCC is the third-commonest etiology of cancer-associated mortality. NAFLD as well as NASH-associated HCC represent the most rapidly escalated indication for liver transplantation. Cirrhosis exists in about 60% of cases of NAFLD as well as NASHassociated HCC.120 This points out that HCC can be stimulated by NAFLD and NASH without cirrhosis. Hence, the belief is that inflammatory parameters will also play a key role in NAFLD or NASH-obtained HCC.
Gut-obtained Endotoxin
Already detailed how gut-obtained endotoxins work in the form of alternative inflammatory parameters and play a significant role in the generation of NAFLD and NASH. The amounts of LPS, alias endotoxins, are further escalated in the portal and peripheral venous veins of patients with HCC.121 They facilitate significantly the invasion potential besides inducing EMT, despite them hampering tumor growth as well.122 LPS stimulates JNK in addition to MAPK through TLR4 in HCC cells, while hampering JNK in addition to MAPK causes a significant reduction in EMT.122 Hence, LPS-TLR4 signaling might be one of the lucrative pathways for controlling the propagation from NAFLD-NASH to HCC.123
Adipokines
Adipokines represent inflammatory parameters associated with HCC generation. Expression of adiponectin in human HCC has an inverse association with tumor size.124 It escalates phosphorylation of JNK and activation of caspase-3, resulting in apoptosis in HCC.124 The hampering of JNK phosphorylation avoids the anti-apoptotic actions of adiponectin.124 Adiponectin has chemoshielding actions besides hepatoshielding actions through sulfatase 2 (SULF2) in HCC.125 Adiponectin deletion facilitates fibrosis and HCC propagation in a choline-deficient NASH mouse model.126 Conversely, high amounts of circulating adiponectin make it feasible to anticipate the subsequent generation of HCC along with poor HCC survival.127 Moreover, adiponectin hampers the oncogenic action of leptin on cell proliferation, migration, and invasion of HCC.31
Leptin expression is increased in hepatoma tissues and cell lines.128 Regulatory T-cells (TRegs), effector CD4+ T-cells, and CD8+T-cells result in stimulation of the expression of the leptin receptor (LEPR) in the liver following the generation of HCC.129 Macrophages and dendritic cells upregulate LEPR expression on T-cells. Leptin hampers activation of TRegs as well as function.127 Escalated leptin expression in HCC is correlated with the expression of human telomerase reverse transcriptase (hTERT).128 Leptin might play a key role in obesity-associated tumorigenesis. Adipokines that include adiponectin along with leptin are critical actors in obesity-associated conditions and might be implicated in the etiopathogenesis of NAFLD and HCC.
UNEXPLORED PATHS
Variable and exciting queries might be there in view of these organokines influencing each other mutually and connecting via different endocrine, autocrine, and paracrine pathways. It has been acknowledged that variable environmental factors, including GM and genetic factors, are associated with the expression of organokines; however, which lifestyle alterations might modulate the expression of these mediators?
Which patients with NASH with a certain extent of therapeutic intervention (diet, physical activity, pharmacologic therapy) vary quantitatively as well as qualitatively in generating these organokines (hepatokines) in contrast to patients with decreased physical activity besides those without undergoing treatment?
Genetic Factors
We have already reviewed how recently the work of researchers aided in illustrating the mode of the risk of cardiovascular disease in sub-kinds of NAFLD (metabolic along with genetic). It has been established that carriers of numerous single nucleotide polymorphisms (SNPs) areas along with mitochondrial mutations possess greater proneness to NAFLD.11 Nevertheless, conferring protection against coronary artery disease (CAD) was illustrated for different SNPs of this type,130 which pointed out that each mutation region might implicate a distinctive mode of NAFLD proneness or CVD avoidance.131 Actually, recent meta-analysis research in the context of NAFLD proneness genes does not result in CAD intrinsically.132 Of the carriers of NAFLD SNPs, a robust association visualized total cholesterol (TC), along with low-density lipoprotein-cholesterol (LDL-C), with CAD, but not plasma TG, along with HDL-C. Enhancement of outflow of FAs as well as “de novo’’ lipogenesis starts the accrual of fat in the liver, besides pushing for more generation of VLDL, which tilts the plasma lipid balance towards a proatherogenic direction along with CVDfacilitating miieu.132 Nevertheless, certain NAFLD-correlated SNPs result in dysfunctional VLDL liberation (TM6F2 as well as PNPLA3,133 as well as microsomal triacylglycerol transfer protein (MTTP), along with phosphatidylethanolamine methyltransferase (PEMT)), hence resulting in a reduction of plasma lipids, with hence the provision of protection conferred to the heart. The genetic variant of PNPLA3, an enzyme encoding I148M (rs738409 C/G) and involved in the hydrolysis of triacylglycerols in adipocytes, has been reported to be associated with NAFLD independent of the metabolic syndrome. Similarly, the genetic variant of the lipid transporter located on the ER (endoplasmic reticulum) and ER-Golgi compartments, TM6SF2 (transmembrane 6 superfamily member 2), encoding E167K (rs58542926 C/T), causes loss of function of the protein and increases hepatic deposition of triglycerides.134 What are the ways by which genetic differences disrupt the generation (expression, formation) of these organokines, hence the proneness to generating metabolic diseases of the liver along with different organs? Recently, we had already emphasized the role of ER stress in T2DM, so no further details were provided.135
Diet as well as Gut Microbiota
The metabolism of certain dietary substances takes place by GM, generating metabolites that have the capacity to modulate the host metabolism. Conversely, diet possesses the capacity to manipulate microbiota diversity and, sequentially, advantageous or inimical actions in the host. These diet disruptions might be restricted to the gut; however, others possess systemic actions resulting in IR and might be associated with MetS risk factors.9,136 Moreover, diet controls certain organokines like FGF21.136
Certain publications have illustrated the association between the microbiome and NAFLD and NASH pathogenesis, apart from the fact that crosstalk between gut and liver (the gutliver axis) plays a key role in the generation of NASH besides its evolution.137 Microbiome-obtained constituents can impact the liver through biomarkers leading to actions that are mutually connected, like gene expression, pro-inflammatory signaling, and manipulations in metabolism leading to toxicity leading to liver inflammation and fibrosis.138
Different assumptions are feasible: a diet disrupts the microbiota, which disrupts the generation of metabolites correlated with MetS (directly correlated with NAFLD or NASH). At the time of liver inflammation, the liberation of organokines (hepatokines) changed; probably crosstalk amongst other organokines changed, an aggravating event in a vicious cycle. Hence the query arises: is it feasible that GM modulates the formation of organokines? What is the way GM disrupts the expression of hepatokines in patients with liver disease? In the case of the gutliver axis, which plays a part in the pathogenesis and propagation of NASH, the maximum probability is that alterations in GM impact NASH generation and indirectly via hepatokines liberation. Since hepatokines take part in the metabolism of other organs and tissues like the pancreas, AT, and muscle, they might be impacted.
Exercise
Certain scientists believe that exercise and the maintenance of muscle mass might modulate circulating quantities of different organokines. Liver steatosis and fibrosis in patients with NAFLD might be avoided independently of weight reduction. The advantages of exercise are apparently correlated with the modulation of crosstalk amongst organs, leading to organokine balance, reduced inflammation, and OS.139
Sleep, Melatonin Relation with Organokines
It is acknowledged that a correlation exists between physical activity and melatonin liberation, which in turn impacts the skeletal muscle makeup (fiber types) and its metabolism. As skeletal muscle liberates myokines and crosstalks with other organs, it is believed that a probable causative association exists between melatonin generation, organokine liberation, and the extent of inflammation. This might be associated with the generation of NAFLD and NASH.
Sleep deprivation and bad sleep quality are considerably responsible for the pathogenesis of metabolic impairment, including NAFLD.140 If NAFLD/NASH is existent, it is feasible that changes in hepatokines liberation will take place. Sequentially, it might change the liberation of other organokines and crosstalk.
Certain publications have assessed the association between melatonin and skeletal muscle metabolism.141 Muscle is acknowledged to liberate myokines with variable metabolic actions. The query initiated is: is there a correlation between melatonin and myokine liberation? If so, can endogenous formation or therapeutic melatonin disturb the generation of organokines? Would it possess any therapeutic advantages? The mode by which sleep quality can disrupt quantitative and qualitative organokine liberation.
Other Metabolic Situations
It is feasible that other situations like iron overload (hyperferritinemia) or dysmetabolic iron overload syndrome can change the generation of these hepatokines, thus causing sequential crosstalks. Does this play a part in the pathogenesis and propagation of NASH (circuit and feedback)?
Management and Treatment
For the NASH generation, the two-hit model is acknowledged. Numerous organokines take part in the 1st or 2nd hit, hence impacting the liver disease initiation. Apart from that, since organokines might be implicated in initiating NAFLD, followed by propagation to NASH, it is plausible to utilize a dose of these biomarkers as a marker of liver disease and propagation.
In the future, we can consider whether there is a probability of pharmacological manipulation of organokine generation. Can we use it as a therapeutic approach for regulating T2D, obesity, MetS, hypertension, CVD and NASH?
CONCLUSION AND FUTURE DIRECTIONS
With the obesity pandemic and subsequently escalated NAFLD cases, the incidence of HCC is not only increasing with NAFLD progressing to cirrhosis but in earlier stages as well. Hence the significance of earlier diagnosis and the avoidance of NAFLD generation. Hence, the group of Yang et al142has generated regulatory networks and crosstalk among various organokines. Yang et al142 further detailed the stellakines generated by HSCs. Moreover, they have developed an ingenuity pathway analysis (IPA) regarding variable organokines. Furthermore, they have found an association between NFκB signaling and variable organokines (Figure 8). On that basis, they have evaluated the role of silymarin in NAFLD and NASH.
Figure 8. NF-κB and Stellakine. IPA Showed the Molecular Pathways of Stellakines and NF-κB Network
Adopted from Yang et al144 APP: amyloid precursor protein, CC chemokines (such as CCL2, CCL11), CSF1: colony-stimulating factor 1 (also known as M-CSF), CTGF: connective tissue growth factor (also called CCN2), CXC chemokines (such as CXCL1, CXCL10, CXCL12, CXCL14, CXCL16), GAS6: growth arrest-specific 6, NTN1: netrin-1, POSTN: periostin. Solid lines indicate direct regulation; dashed lines indicate indirect regulation.
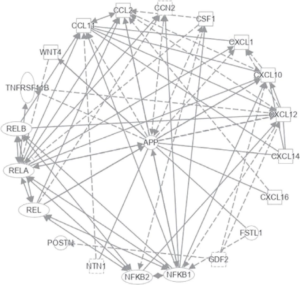
Activated HSCs secrete numerous pro-inflammatory factors and generate ECM components like α-SMA, type I and type III collagen fibers, which guide liver inflammation and modulate hepatocyte demise, hence facilitating the generation of liver disease.
The four main flavonolignan isomers in silymarin are silybin, isosilybin, silymarin, and silybin. Silymarin has antioxidant properties, anti-inflammatory properties, anti-fibrotic actions, and IR manipulation.
Its mode of action, silymarin, escalates the generation of glutathione in the liver by escalating the accessibility of cysteine, its biosynthetic substrate, which in turn aids in escalating its antioxidant ability in liver tissue. The main modes of action of silymarin in protecting liver cells are: (1) stabilization of membrane permeability by hampering lipid peroxidation and aiding the liver in the sustenance of quantities of glutathione; (2) silymarin also blocks the activation of NF-κB by hampering the formation of TNF-α, interferon-γ, IL-2, and IL-4, thus avoiding various actions of toxic chemicals like carbon (3) It further hampers the expression of TNF-stimulated by α-amanita toxin of the poisonous mushroom, etc.143
Of the various trials of silymarin conducted in Malaysia, the USA, and Pakistan, finally, Phase III clinical trials have validated that silymarin is the most beneficial drug for NAFLD patients currently; however, its standardization, dosage form, and dosage regimen are tough problems to overcome now. Together, these outcomes point out that silymarin treatment is efficacious in decreasing fibrosis, liver stiffness, and metabolic markers (alanine transaminase (ALT) and aspartate transaminase ((AST)); improving OS and endothelial impairment; and postponing disease propagation invariable areas. Silymarin is anticipated to be an efficacious therapeutic drug for NAFLD and NASH (Figure 8).144 Hence, greater research on these organokines regarding NAFLD and NASH would be followed by more therapeutic targets by targeting any of these organokines to be able to avoid deaths in young subjects even in early NAFLD from HCC.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.


