INTRODUCTION
Iron (Fe) and copper (Cu) are bioelements and vital transition metals whose deficiency or excess in the organism are associated with pathologic situations. Both metals are clearly hormetic, they are required at low levels for human health (Recommended Daily Intake: 10-15 mg Fe/ day and 1-3 mg Cu/day) but at higher levels (more than 30 mg Fe/day or 8 mg Cu/day) they produce toxic effects in liver and brain.1-4 The metal toxicity seems due to their participation in the Haber-Weiss redox reaction that produces the highly reactive HO•.5,6 The intracellular steady-state concentrations of reactive oxygen species indicate that H2O2 and ROOH are the quantitatively predominant species by a factor of 104-1011.7
Currently, there are two hypotheses for the molecular mechanism of transition metal toxicity in mammalian organs. The two hypotheses are not incompatible and it is likely that the two processes occur simultaneously. The first one considers that the reduced forms Fe2+ and Cu+catalyze the homolytic scission of the O-O bond in H2O2 and ROOH in a Fenton-like reaction to produce HO• and RO• radicals. The second one considers the reaction of Fe3+ and Cu2+ with intracellular reduced glutathione (GSH), due to the high affinity of the two metals ions for the thiol (-SH) group. This depletes cells of GSH that is equilibrated with essential thiol groups in enzymes and regulatory factors. Concerning the first hypothesis, there are two points of view of
the O-O bond homolysis: a classical one where Fe2+ or Cu+ directly catalyzes the reaction and a second one, in which Fe2+ and Cu+
bind to a specific peptide or protein that reacts with H2O2 generating HO•, that is able to oxidize neighboring amino acids and to produce protein crosslinking, fragmentation and denaturation.1,2,8 The last mechanism seems to apply to β-amyloid in Alzheimer’s disease.9
The toxicological effects of Fe and Cu overloads were studied in rat liver by a kinetic and holistic approach, considering the time (t1/2) and the metal liver content (C50) for half-maximal effects. The kinetic approach refers to the central role of the t1/2 to define the sequence of events in the liver, and the holistic concept considers the whole organ, as in liver chemiluminescence and homogenate determinations.
TOXICITY PROCESSES IN Fe AND Cu OVERLOADS
Sprague Dawley male rats (200-230 g) received i.p. (a) for Fe t1/2 determination, 30 mg/kg rat of ferrous chloride (FeCl2.4H2O), that corresponds to 8.42 mg Fe element/kg; (b) for Fe C50 determination, 5-60 mg/kg of ferrous chloride, corresponding to 4.1-49.2 mg/kg of Fe element/kg; (c) for Cu t 1/2 determination, 10 mg/kg rat of cupric sulfate (CuSO4 .5H2O), that corresponds to 2.54 mg/kg of Cu element/kg; and (d) for Cu C50 determination, 3-30 mg/kg of cupric sulfate, corresponding to 0.763-7.63 mg/kg of Cu element. Control rats received a similar volume of saline solution.
The effects of Fe and Cu intoxications in rat liver are summarized in Table 1. Both metals produced a situation of oxidative stress, that was originally described as an unbalance between oxidant production and antioxidant defense.10 The concept has been updated and now considers that oxidative reactions lead to a disruption of redox signaling and control, and to molecular and cellular damage.11 A high content of cellular–SH groups is considered essential for cell regulation and survival.
The description of the metal effects in rat liver given in Table 1, is based on the time (t1/2) and on the metal liver content (C50) for half maximal effects. The t1/2 describes the kinetics of the effect at the used doses. At variance, C50 establishes the effect concentration dependence.
| Table 1: Rat Liver Oxidative Stress after Fe and Cu Overloads.a |
|
Property |
Fe |
Cu |
| Effect (%) b |
t1/2 (h) |
C50 (µg Fe/g) c |
Change (%) |
t 1/2 (h) |
C50 (µg Cu/g) c |
| Rat survival |
(-) 10 |
About 240 |
140 |
(-) 40 |
About 120 |
120 |
| Metal content |
(+) 40 |
4 |
60 |
(+) 1000 |
5 |
80 |
| Liver chemiluminescence |
(+) 200 |
4 |
115 |
(+) 100 |
4 |
42 |
| Lipoperoxidation |
(+) 200 |
6.5 |
130 |
(+) 100 |
7 |
45 |
| Protein oxidation |
(+) 60 |
4 |
116 |
(+) 40 |
5.5 |
50 |
| Hydrophilic antioxidant |
(-) 60 |
5 |
118 |
(-) 75 |
4 |
42 |
| Hydrophobic antioxidant |
(-) 50 |
5 |
120 |
(-) 50 |
5 |
52 |
| GSH content |
(-) 58 |
4 |
116 |
(-) 79 |
4 |
40 |
| GSH/GSSG ratio |
(-) 50 |
2 |
108 |
(-) 50 |
2 |
30 |
| SOD1 activity |
(+) 57 |
8 |
114 |
(+) 127 |
8.5 |
42 |
| Catalase activity |
(+) 65 |
8.5 |
110 |
(-) 26 |
8 |
44 |
| GPx activity |
(-) 39 |
4.5 |
120 |
(-) 22 |
5 |
48 |
| a Adapted from references. (1-3): b in % of increased (+) or decreased (-) property compared with control rats; c determined by atomic absorption. |
In Fe overload, 90% of the rats survived the observation time of 48 h, whereas in Cu overload, 85% of the animals survived 24 h and 60% survived 48 h.3 Metal accumulation in the liver was dose- and time-dependent.1 The liver content of Fe increased 1.6 times and the one of Cu 11-fold after 48 h of metal overloads. The Fe and Cu contents observed in rat liver are similar to those in patients with haemochromatosis12 or with Wilson’s disease, respectively.13
LIVER OXIDATIVE STRESS AND DAMAGE
The increased intracellular levels of Fe2+ and Cu1 (Figure 1) lead to an enhanced homolytic cleavage of H2O2 yielding HO• and initiating phospholipid peroxidation and protein oxidation. The spontaneous light emission from in situ mammalian organs is a physiological phenomenon and also an assay for the determination of the rate of lipid peroxidation, from singlet oxygen (1O2) steady states.14 The molecular mechanism of light emission includes the Russell’s reaction in which two secondary or tertiary peroxyl radicals (ROO•) yield 1O2 or excited carbonyl groups (>C=O*) as products and the 1O2 dimol emission. Two 1O2 molecules upon collision produce a photon at 634 or 703 nm, whereas >C=O* yields photons at 460-470 nm.14
Figure 1: Scheme of the Biochemical Process in Fe and Cu Liver Toxicity.1
In red, effects of Fe toxicity and in Blue, effects of Cu toxicity.
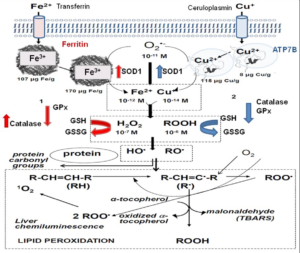
Increased liver chemiluminescence indicates an enhanced rate of free-radical mediated lipid peroxidation. This process was also monitored by the determination of the lipid peroxidation product TBARS. Similarly, carbonyl groups, >C=O were determined as products of protein oxidation. The indicators of oxidative reactions and damage (chemiluminescence, TBARS, and >C=O) exhibited about similar t1/2, considering the experimental error.
Antioxidants are enzymes or small molecules that decrease the level of oxidative chemical species. The small molecules able to trap free- radicals and to reduce the extent of phospholipid peroxidation and of protein oxidation, are GSH (mM range), α-tocopherol and β-carotene (μM range). Other kind of antioxidants, more important from a physiological consideration, are the enzymes Cu, Zn-superoxide dismutase (SOD1), catalase, and glutathione peroxidase (GPx).
Rat liver antioxidant defenses were affected by Fe and Cu overloads. Hydrophilic antioxidants were decreased, measured either as a pool, or as GSH concentration. Similarly, hydrophobic antioxidants were diminished. The decrease of both types of antioxidants indicates antioxidant consumption, consistent with an increased rate of liver oxidative free-radical reactions. Antioxidant consumption exhibited similar t1/2, coincident with the idea that the observed oxidative damage is due to a common free-radical mediated mechanism.
The two main antioxidant enzymes, SOD and catalase, evolved with aerobic life in bacteria.15 Animal biochemistry kept this ancestral defense mechanism and SOD1 and catalase decreased activities have been associated with pathological conditions in mammals and humans.6,16 The adaptive response of increased SOD1 and catalase activities in mammalian organs was early reported in neonatal rabbit lung,17 and recognized as a strategy of antioxidant defense in mammals18 and in humans.19 In Fe overload, there was an increase in SOD1 and catalase activities in response to oxidative stress. In Cu toxicity, a different response was observed in the two enzymes: SOD activity increased but catalase activity decreased. GPx activity was decreased after Fe and Cu overloads, it is likely that increased phospholipid peroxidation would include high levels of ROO• (peroxyl radical) that binds to the enzyme and inactivates the reaction center.
The adaptive response involving the described increased enzyme activities is likely to be mediated by the nuclear factor erythroid 2- elated factor 2 (Nrf2) transcription factor that appears as responsive to increases in ROOH intracellular levels.5
The mitochondrial respiration of rat liver mitochondria isolated from Cu-overloaded rats showed significant decreases in the active ATP-forming state 3, but not in the resting state 4. With malate-glutamate as substrate, state 3 respiration was 36% diminished, and with succinate, O2 uptake was 25% decreased (Table 2).
| Table 2: Oxygen Uptake of Rat Liver Mitochondria after Cu Overload. a |
|
Oxygen uptake (ng-at O/min × mg protein) |
| Control |
Cu treated |
| Substrate: 5 mM malate, 5 mM glutamate |
| State 3 |
47±2 |
30±3* |
| State 4 |
6.4±0.4 |
5.0±0.3 |
| Respiratory control |
7.3±0.1 |
6.0±0.2 |
| Substrate; 10 mM succinate |
| State 3 |
64±3 |
48±3* |
| State 4 |
11.4±0.6 |
10.2±0.5 |
| Respiratory control |
4.5±0.1 |
4.7±0.1 |
| a Rats received 25.5 mg/kg of cupric sulfate/kg rat, corresponding to 6.5 mg of Cu element /kg rat, 6 h before rat sacrifice. *p<0.05. |
CONCLUSION
Rats exposed to Fe and Cu overloads develop liver oxidative stress in which cells may adapt to the situation or succumb with eventual cell death. The adaptation includes the up-regulation of SOD1 and catalase synthesis and of enzymes involved in GSH conjugation and in GSH synthesis. The apparent purpose of the adaptive response is to overcome the oxidative challenge and to restore reactive oxygen species to levels compatible with cell life. The molecular mechanisms underlying cell adaptation are at present not fully understood. However, in recent years some transcription factors, as Nrf2, have emerged as master regulators of the adaptive response. Decreased liver GSH, consequence of Fe and Cu accumulation and the ensuing oxidative stress, trigger downstream signaling as an attempt to keep the normal composition of membranes and proteins. Given its high intracellular concentration in liver (5-7 mM), GSH defines the cellular redox potential and protects cells against oxidative stress. The whole process of metal toxicity is constituted by oxidative biochemical processes with t1/2 of 4.6±0.5 h for Fe and of 4.9±0.6 h for Cu, that encompass increased free-radical mediated oxidations and decreased GSH contents, superimposed with the adaptive response of the antioxidant enzymes. Altogether, this set of biochemical changes appears to indicate an oxidative stress situation where cells are unable to control the enhanced production of oxidative species.
DECLARATION OF INTEREST
The authors declare that they have no competing interests.


